

Key Points
Sera from COVID-19 patients induce increased thrombus formation.
cAMP elevation prevents antibody-induced procoagulant PLT generation and thrombus formation.

Abstract
Thromboembolic events are frequently reported in patients infected with the SARS-CoV-2 virus. The exact mechanisms of COVID-19-associated hypercoagulopathy, however, remain elusive. Recently, we observed that platelets (PLTs) from patients with severe COVID-19 infection express high levels of procoagulant markers, which were found to be associated with increased risk for thrombosis. In the current study, we investigated the time course as well as the mechanisms leading to procoagulant PLTs in COVID-19. Our study demonstrates the presence of PLT-reactive IgG antibodies that induce marked changes in PLTs in terms of increased inner-mitochondrial transmembrane potential (Δψ) depolarization, phosphatidylserine (PS) externalization, and P-selectin expression. The IgG-induced procoagulant PLTs and increased thrombus formation were mediated by ligation of PLT Fc-γ RIIA (FcγRIIA). In addition, contents of calcium and cyclic-adenosine-monophosphate (cAMP) in PLTs were identified to play a central role in antibody-induced procoagulant PLT formation. Most importantly, antibody-induced procoagulant events, as well as increased thrombus formation in severe COVID-19, were inhibited by Iloprost, a clinically approved therapeutic agent that increases the intracellular cAMP levels in PLTs. Our data indicate that upregulation of cAMP could be a potential therapeutic target to prevent antibody-mediated coagulopathy in COVID-19 disease.
Introduction
Infection with SARS-CoV-2 is associated with abnormalities in the coagulation system, with an increased incidence of thromboembolic events in small vessels leading to higher mortality.1-3 Upregulated release of inflammatory cytokines and increased interactions between different actors of innate and adaptive immunity have been suggested to be the main causes for the prothrombotic environment observed in COVID-19 disease.4 In addition, a significant number of reports described platelet (PLT) hyperactivity in patients with COVID-19.5,6 Procoagulant PLTs, predominantly generated at the outer side of the growing thrombus, are increasingly recognized to link primary with secondary hemostasis.7-10 The latter is mediated by negatively charged membrane phospholipids externalized on procoagulant PLT surfaces. This unique feature of procoagulant PLTs enables the assembly of tenase as well as prothrombinase complexes, leading to high thrombin burst, increased fibrin deposition, and thrombus formation.11 Recently, we showed that PLTs from patients with severe COVID-19 infection express procoagulant phenotype. Immunoglobulin G (IgG) fractions were found to be responsible for the COVID-19-associated procoagulant PLTs.12
In the current study, we investigated the time course of the generation of antibody-induced procoagulant PLTs as well as the underlying mechanisms leading to alterations in PLT phenotype in COVID-19. We observed that IgG fractions from severe COVID-19 patients induce increased thrombus formation in Fc-γ RIIA (FcγRIIA)-dependent manner. More importantly, cyclic-adenosine-monophosphate (cAMP) elevation prevented antibody-induced procoagulant PLT generation as well as thrombus formation.
Methods
Study design
Experiments were performed using leftover serum material from intensive care unit (ICU) COVID-19 patients who were referred to our laboratory between March and June 2020. Some of these cases have been reported in Althaus et al. All experiments presented in this study were performed independently, and no overlay in the results exists. The diagnosis of SARS-CoV-2 infection was confirmed by real-time polymerase chain reaction (PCR) on material collected by nasal swabs. To consider unspecific ICU effects on PLTs, an ICU non-COVID-19 patient control group was enrolled. Additionally, sera were collected from healthy blood donors at the Blood Donation Centre Tuebingen after written consensus was obtained to establish cutoff values when appropriate. When indicated, IgG fractions were isolated from the corresponding sera using a commercially available IgG purification kit (MelonTM-Gel IgG Spin Purification Kit, Thermo Fisher Scientific, Waltham, MA). Additional details are available in the supplemental Data.
Detection of COVID-19 antibody-induced effects
Washed platelets (wPLTs) were prepared from venous blood samples as described previously13 and incubated with serum/IgG from ICU COVID-19 patients or controls. Changes in the inner mitochondrial transmembrane potential (ΔΨ), phosphatidylserine (PS) externalization, P-selectin (CD62p), and glycoprotein VI (GPVI) expression on wPLTs were analyzed by flow cytometry (FC). Additional information is provided in the supplemental Data.
Assessment of thrombin generation (TG) and in vitro thrombus formation
ICU COVID-19-induced TG was tested using calibrated automated thrombogram (CAT; Stago, Maastricht, Netherlands) according to the manufactureŕs instructions. To assess the impact of ICU COVID-19 IgG-induced effects on thrombus formation, an ex vivo model of thrombus formation utilizing hirudin as well as recalcified citrated blood was established. A microfluidic system (BioFlux 200, Fluxion Biosciences, Alameda, CA) was used at a shear rate of 1500−1 (60 dyne) according to the recommendations of the International Society on Thrombosis and Haemostasis (ISTH) standardization committee for biorheology.14 Additional information is provided in the supplemental Data.
Statistics
Statistical analyses were performed using GraphPad Prism 7 (La Jolla, CA). Student t test was used to analyze normally distributed results. A nonparametric test (Mann-Whitney U test) was used when data failed to follow a normal distribution as assessed by D’Agostino and Pearson omnibus normality test. Group comparison was performed using the Wilcoxon matched-pairs signed-rank test and the Fisher exact test with categorical variables. A P value < .05 was assumed to represent statistical significance.
Ethics
Studies involving human material were approved by the Ethics Committee of the Medical Faculty, Eberhard-Karls University of Tuebingen, Germany, and were conducted in accordance with the Declaration of Helsinki.
Results
Patient characteristics
Patients were enrolled in this study between 1 March 2020 and 16 June 2020. Thirty sera of patients with confirmed severe SARS-CoV-2 infection were admitted to the ICU (patient numbers 1 to 30; Figure 1; supplemental Table 1). Clinical data from 21 of these ICU COVID-19 patients were reported in a previous study.12 The mean age of ICU COVID-19 patients was 58 years (range: 29 to 88 years). 20 of 30 (67%) patients had known risk factors for severe COVID-19 infection as described previously,15 including hypertension (18 of 30; 60%), obesity (6 of 30; 20%), coronary artery disease (4 of 30; 13%), and diabetes mellitus (6 of 30; 20%). Elevated D-Dimer levels were detected in all patients (median, range: 3.4 mg/dL, 0.9 to 45.0 mg/dL) and thrombosis was diagnosed in 13 of 30 (43%) patients. Longitudinal blood samples were available from 4 COVID-19 patients (patient numbers 27 to 30) who were first admitted to the normal ward and later to the ICU for mechanical ventilation. As an ICU control group, 5 patients who were admitted to the ICU due to non-COVID-19 related causes were included in this study (supplemental Table 1).
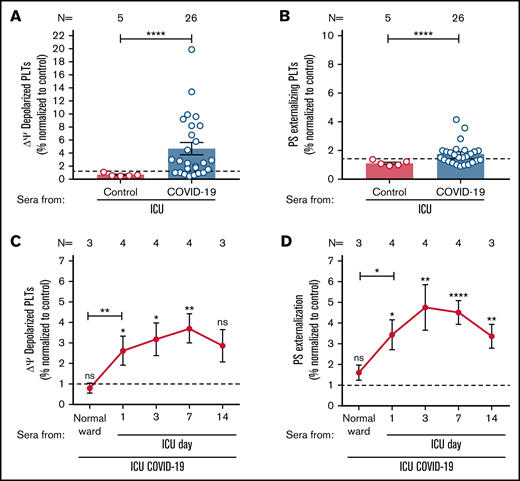
ICU COVID-19 patient serum-induced effects on PLTs during disease. (A-B) ICU COVID-19 (n = 26) or ICU non-COVID-19 control (n = 5) patient serum-induced changes in Δψ as well as PS externalization in wPLTs were analyzed by FC. (C-D) Sera of 4 ICU COVID-19 patients were collected for up to 14 days during hospitalization and analyzed for their ability to induce changes in Δψ as well as PS externalization in wPLTs via FC. Data are presented as mean ± SEM of the measured fold increase (FI) compared with control. The number of patient sera tested is reported in each graph. Dot lines in (A,B) represent the calculated cutoffs determined by testing sera from healthy donors as mean of FI + 2 × SEM. *P < .05, **P < .01, ***P < .001, and ****P < .0001. ns, not significant; FC, flow cytometry; HC, healthy control; Δψ, inner mitochondrial transmembrane potential; N, number of HCs or patients; PS, phosphatidylserine.
ICU COVID-19 patient serum-induced effects on PLTs during disease. (A-B) ICU COVID-19 (n = 26) or ICU non-COVID-19 control (n = 5) patient serum-induced changes in Δψ as well as PS externalization in wPLTs were analyzed by FC. (C-D) Sera of 4 ICU COVID-19 patients were collected for up to 14 days during hospitalization and analyzed for their ability to induce changes in Δψ as well as PS externalization in wPLTs via FC. Data are presented as mean ± SEM of the measured fold increase (FI) compared with control. The number of patient sera tested is reported in each graph. Dot lines in (A,B) represent the calculated cutoffs determined by testing sera from healthy donors as mean of FI + 2 × SEM. *P < .05, **P < .01, ***P < .001, and ****P < .0001. ns, not significant; FC, flow cytometry; HC, healthy control; Δψ, inner mitochondrial transmembrane potential; N, number of HCs or patients; PS, phosphatidylserine.
Sera from ICU COVID-19 patients induce an increase in procoagulant PLTs
To investigate whether sera of ICU COVID-19 patients have the potential to induce an increased Δψ depolarization as well as PS externalization on the PLT surface, wPLTs from healthy individuals were incubated with sera from 26 ICU COVID-19 patients with a severe course of disease as well as 5 ICU non-COVID-19 patients. Based on the calculated cutoffs (mean + 2 × SD of healthy controls [HCs]), 19 of 26 (73%) sera from patients with severe COVID-19 disease induced significantly higher Δψ depolarization in PLTs from healthy donors compared with ICU controls (fold increase [FI] in percent Δψ depolarization ± SEM: 6.10 ± 1.12 vs 0.67 ± 0.10, P value < .0001; Figure 1A). In addition, significantly higher PS externalization was observed when PLTs were incubated with ICU COVID-19 sera compared with ICU control sera (FI in percent PS ± SEM: 2.12 ± 0.19 vs 1.12 ± 0.08, P value < .0001; Figure 1B).
Next, we sought to investigate the time course of the observed changes in both markers. Longitudinal blood samples were available from 4 ICU COVID-19 patients. Sera were collected at hospital admission (normal ward or ICU) and during a follow-up period at ICU for up to 14 days. As shown in Figure 1C-D, sera from ICU COVID-19 patients induced significant changes in Δψ depolarization and PS externalization as clinical manifestation worsened, requiring admission to ICU. Of note, serum-induced changes peaked within day 3 and day 7 of the ICU stay (FI in percent Δψ depolarization ± SEM: 3.71 ± 0.72, P value = .0070; and percent PS externalization ± SEM: 4.80 ± 1.11, P value < .0001, respectively; Figure 1C-D; supplemental Figure 1). Notably, the rise of PLT markers was associated with increasing levels of detected IgGs against the spike S protein of SARS-CoV-2 in the corresponding ICU COVID-19 patients’ follow up sera but not in the total IgG contents of isolated IgG fractions (supplemental Figure 2A-B). Concerned that ICU COVID-19 serum induced PLTs effects might be due to the potential prevalence of immune complexes (ICs) on the PLT surface, we performed IC removal by absorbing sera with polyethylene glycol (PEG). Interestingly, ICU COVID-19-induced effects remained unchanged in IC absorbed patient sera (supplemental Figure 3).
IgGs from severe COVID-19 trigger procoagulant PLTs, GPVI shedding, thrombin generation, and increased ability to form thrombus
To further verify the impact of sera from severe COVID-19 patients on PLTs, the expression of the α-granule release and PLT activation marker CD62p was analyzed in double staining in parallel to PS. Additionally, the expression of PLT GPVI was investigated following IgG incubation. FC analyses revealed that IgG fractions from severe COVID-19 patients induce remarkable changes in the distribution of CD62p/PS positivity. In contrast, the PLT population was almost nonaffected after incubation with IgGs from HCs or ICU non-COVID-19 control patients. Overall, higher percentage of double-positive events was observed after incubation with IgGs from ICU COVID-19 patients compared with ICU and to HCs (percent CD62p/PS-positive PLTs ± SEM: 31.63 ± 3.86 vs 4.04 ± 1.16, P value = .0007; and vs 2.88 ± 0.52, P value < .0001, respectively; Figure 2A; supplemental Figure 4). Moreover, analyses for PLT GPVI expression revealed that ICU COVID-19 IgGs have the capability to induce significant shedding of the collagen receptor on the PLT surface, whereas these effects were nearly absent in ICU non-COVID-19 and HC IgG incubated PLTs (mean percent GPVI negative PLTs ± SEM: 67.66 ± 7.14 vs 6.68 ± 1.60, P value = .0043; and vs 7.24 ± 1.00, P value = .0043, respectively; Figure 2B). Interestingly, ICU COVID-19 IgG also showed the potential to reduce expression levels of the antiapoptotic protein BCL-XL, whereas these changes were absent in HC IgG incubated PLTs (supplemental Figure 5).

ICU COVID-19 IgGs induce procoagulant PLTs, GPVI cleavage, increased thrombin generation and thrombus formation on collagen. (A) HC, ICU non-COVID-19, and ICU COVID-19 IgG-induced changes in wPLTs externalization of PS and expression of CD62p were analyzed via annexin V-FITC and CD62p-APC antibody staining, respectively. Data are shown as mean percentage ± SEM of annexin V-FITC and or CD62p-APC-positive-labeled wPLTs. (B) ICU-COVID-19 IgG-induced reductions in the expression of GPVI on the PLT surface were analyzed by GPVI-PE antibody staining and compared as percentage of GPVI-negative PLTs ± SEM to the controls. (C) PRP from healthy individuals was preincubated with HC or ICU COVID-19 IgG and analyzed for thrombin generation using CAT. Each curve represents the amounts of generated thrombin over time induced by HC (dotted blue line) or IgG from different ICU COVID-19 patients (red line) (D) PRP from healthy individuals with the blood group O was incubated with HC, ICU non-COVID-19 control, or ICU COVID-19 IgG, labeled with FITC conjugated calcein and perfused through microfluidic channels at a shear rate of 1500−1 (60 dyne) for 5 minutes after reconstitution into autologous whole blood. Images were acquired at x20 magnification in the fluorescent (upper panel) as well as in the BF channel (lower panel). Scale bar 50 µm. (E) Mean percentage of surface are covered (SAC) by thrombus ± SEM in the presence of HC, ICU non-COVID-19 control, and ICU COVID-19 IgG after 5 minutes perfusion time. The number of patients and healthy donors tested is reported in each graph. See Figure 1 for P values and abbreviation definitions.
ICU COVID-19 IgGs induce procoagulant PLTs, GPVI cleavage, increased thrombin generation and thrombus formation on collagen. (A) HC, ICU non-COVID-19, and ICU COVID-19 IgG-induced changes in wPLTs externalization of PS and expression of CD62p were analyzed via annexin V-FITC and CD62p-APC antibody staining, respectively. Data are shown as mean percentage ± SEM of annexin V-FITC and or CD62p-APC-positive-labeled wPLTs. (B) ICU-COVID-19 IgG-induced reductions in the expression of GPVI on the PLT surface were analyzed by GPVI-PE antibody staining and compared as percentage of GPVI-negative PLTs ± SEM to the controls. (C) PRP from healthy individuals was preincubated with HC or ICU COVID-19 IgG and analyzed for thrombin generation using CAT. Each curve represents the amounts of generated thrombin over time induced by HC (dotted blue line) or IgG from different ICU COVID-19 patients (red line) (D) PRP from healthy individuals with the blood group O was incubated with HC, ICU non-COVID-19 control, or ICU COVID-19 IgG, labeled with FITC conjugated calcein and perfused through microfluidic channels at a shear rate of 1500−1 (60 dyne) for 5 minutes after reconstitution into autologous whole blood. Images were acquired at x20 magnification in the fluorescent (upper panel) as well as in the BF channel (lower panel). Scale bar 50 µm. (E) Mean percentage of surface are covered (SAC) by thrombus ± SEM in the presence of HC, ICU non-COVID-19 control, and ICU COVID-19 IgG after 5 minutes perfusion time. The number of patients and healthy donors tested is reported in each graph. See Figure 1 for P values and abbreviation definitions.
According to these findings, we were interested in whether ICU COVID-19 antibody-induced PLT changes might affect PLTs’ endogenous thrombin generation potential. Using a thrombin generation assay (CAT), we detected remarkable increases in thrombin generation of PLTs that were preincubated with IgG from ICU COVID-19 patients. We tested the 4 sera that induced an increase in CD62p/PS expression from which we had sufficient volume for thrombin generation assessment. Notably, ICU COVID-19 IgG preincubated PLTs obtained higher peaks of thrombin and a higher velocity index compared with HC IgG (Figure 2C; supplemental Table 2).
Next, we sought to investigate the ability of ICU COVID-19 IgG fractions to cause increased thrombus formation. PLTs from healthy individuals were incubated with IgGs from ICU COVID-19 patients or ICU controls and HCs, added to autologous whole blood samples and finally perfused through collagen-covered microfluidic channels at a shear rate of 1500 s−1 (60 dyne). As shown in Figure 2D, IgG from severe COVID-19 patients caused increased thrombus formation. Overall, significantly higher surface area coverage (SAC) by thrombus was observed in the presence of ICU COVID-19 IgGs compared with ICU controls and HCs (mean percent SAC ± SEM: 13.95 ± 1.55 vs 2.86 ± 1.10, P value = .0070; and vs 2.70 ± 0.83, P value = .0002, respectively; Figure 2E). Notably, similar findings were observed in recalcified citrated blood under restored coagulation conditions (supplemental Figure 6A-B). Together with the increased percentage of procoagulant PLTs (CD62p/PS-positive) and the enhanced potential to form thrombin in response to ICU COVID-19 IgGs, these findings provide a potential explanation for the increased thromboembolic events in severe COVID-19 patients.
ICU COVID-19 IgGs cause procoagulant PLTs via crosslinking FcγRIIA
To further determine the underlying mechanistic pathways leading to ICU COVID-19 IgG-induced formation of procoagulant PLTs, we next considered a potential ligation of FcγRIIA by patients’ sera/IgGs. For this purpose, wPLTs were pretreated with the monoclonal antibody (moAb) IV.3. This FcγRIIA blockade resulted in significant inhibition of the antibody-induced Δψ depolarization (percent Δψ depolarized PLTs ± SEM: 26.80 ± 5.50 vs 5.65 ± 0.34, P value = .0020; Figure 3A). Intriguingly, the blockade of FcγRIIA markedly reduced CD62p/PS double-positive PLT population compared with isotype-control (percent CD62p/PS-positive PLTs ± SEM: 48.91 ± 3.05 vs 12.88 ± 1.65, P value = .0078; Figure 3B; supplemental Figure 7). To further verify the dependency on PLT FcγRIIA in ICU COVID-19 IgG-induced procoagulant PLT formation, PLTs were incubated with corresponding ICU COVID-19 F(ab’)2. Of note, F(ab’)2 that were prepared from procoagulant PLTs inducing ICU COVID-19 IgGs were unable to induce the formation of CD62p/PS-positive PLTs (percent CD62p/PS-positive PLTs ± SEM: 28.32 ± 5.90 vs 2.69 ± 0.94, P value = .0128; supplemental Figure 8).
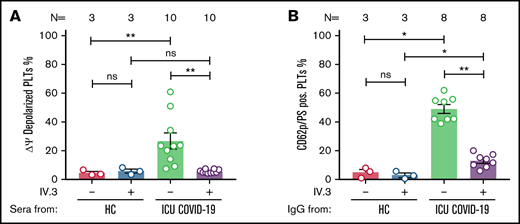
ICU COVID-19 IgG-induced formation of procoagulant PLTs is FcγRIIA dependent. (A) ICU COVID-19 patient serum-induced changes of Δψ as well as ICU COVID-19 IgG-induced formation (B) of procoagulant PLTs were analyzed in the presence or absence of moAb IV.3 via FC. Data are presented as mean percentage of (A) Δψ depolarized PLTs and (B) CD62p/PS-positive PLTs ± SEM. The number of patients and healthy donors tested is reported in each graph. See Figure 1 for P values and abbreviation definitions.
ICU COVID-19 IgG-induced formation of procoagulant PLTs is FcγRIIA dependent. (A) ICU COVID-19 patient serum-induced changes of Δψ as well as ICU COVID-19 IgG-induced formation (B) of procoagulant PLTs were analyzed in the presence or absence of moAb IV.3 via FC. Data are presented as mean percentage of (A) Δψ depolarized PLTs and (B) CD62p/PS-positive PLTs ± SEM. The number of patients and healthy donors tested is reported in each graph. See Figure 1 for P values and abbreviation definitions.
Next, we analyzed the impact of FcγRIIA blockade on antibody-mediated thrombus formation. Pretreatment of PLTs with moAb IV.3 prior to ICU COVID-19 IgG incubation resulted in a significant reduction of thrombus formation compared with isotype-control (mean percent SAC ± SEM: 16.49 ± 1.02 vs 5.84 ± 1.93, respectively, P value = .0090; Figure 4A-B). These results indicate that a subgroup of ICU COVID-19 IgG antibodies has the capability to induce the formation of procoagulant PLTs with increased thrombogenic ability via crosslinking FcγRIIA.
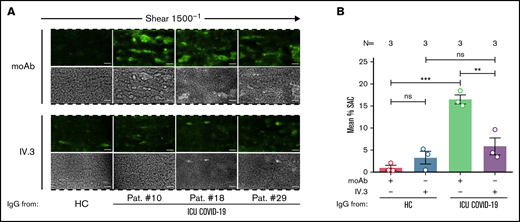
FcγRIIA inhibition prevents ICU COVID-19 IgG-induced thrombus formation. (A) PRP from healthy individuals with the blood group O was incubated with HC or ICU COVID-19 IgG in the presence of moAb IV.3 or isotype control (moAb) and perfused through microfluidic channels at a shear rate of 1500−1 (60 dyne) for 5 minutes. Images were acquired at x20 magnification in fluorescent (upper panel) as well as in the BF channel (lower panel). Scale bar 50 µm. (B) Mean percent of SAC ± SEM induced by HC or ICU COVID-19 IgG in the presence or absence of moAb IV.3 or isotype control (moAb). The number of patients and healthy donors tested is reported in each graph. See Figure 1 for P values and abbreviation definitions.
FcγRIIA inhibition prevents ICU COVID-19 IgG-induced thrombus formation. (A) PRP from healthy individuals with the blood group O was incubated with HC or ICU COVID-19 IgG in the presence of moAb IV.3 or isotype control (moAb) and perfused through microfluidic channels at a shear rate of 1500−1 (60 dyne) for 5 minutes. Images were acquired at x20 magnification in fluorescent (upper panel) as well as in the BF channel (lower panel). Scale bar 50 µm. (B) Mean percent of SAC ± SEM induced by HC or ICU COVID-19 IgG in the presence or absence of moAb IV.3 or isotype control (moAb). The number of patients and healthy donors tested is reported in each graph. See Figure 1 for P values and abbreviation definitions.
Calcium is pivotal for the generation of IgG-induced procoagulant PLTs
Following the detection of an ICU COVID-19 IgG-induced procoagulant PLT phenotype, we sought to investigate the underlying intracellular molecular mechanisms. The contribution of calcium was analyzed by the depletion of extra and intracellular calcium contents via EGTA and 1,2-Bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid tetrakis(acetoxymethyl) ester (BAPTA), respectively. Extracellular calcium depletion caused significant inhibition of Δψ depolarization (percent Δψ depolarized PLTs ± SEM: 52.63 ± 3.12 vs 31.99 ± 3.31, P value = .0079; Figure 5A) as well as the generation of CD62p/PS-positive PLTs (percent CD62p/PS-positive PLTs ± SEM: 32.89 ± 2.77 vs 6.42 ± 1.21, P value = .0039; Figure 5B; supplemental Figure 9).
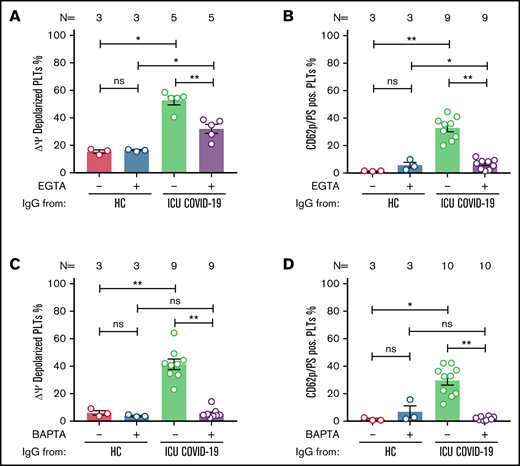
ICU COVID-19 IgG-induced formation of procoagulant PLTs is dependent on calcium. Panel (A-B) shows ICU COVID-19 IgG-induced changes of different PLT markers in the presence (vehicle) or absence of extracellular calcium (EGTA 1 mM). FC detected changes of (A) PLT Δψ and (B) formation of CD62p/PS-positive PLTs after ICU COVID-19 IgG incubation in vehicle or EGTA pretreated wPLTs, respectively. (C-D) ICU COVID-19 IgG-induced PLT changes in the presence (vehicle) or in intracellular calcium depleted (BAPTA 20 µM) wPLTs. FC detected changes of (C) PLT Δψ and (D) formation of CD62p/PS-positive PLTs induced by ICU COVID-19 IgG in vehicle or BAPTA preloaded wPLTs, respectively. Data are presented as mean percentage ± SEM of (A,C) Δψ depolarized PLTs and mean percentage ± SEM of (B,D) PS (Lactadherin-FITC) and CD62p-APC-positive wPLTs. Note that lactadherine is a calcium-independent marker of PS externalization. The number of patients and healthy donors tested is reported in each graph. See Figure 1 for P values and abbreviation definitions.
ICU COVID-19 IgG-induced formation of procoagulant PLTs is dependent on calcium. Panel (A-B) shows ICU COVID-19 IgG-induced changes of different PLT markers in the presence (vehicle) or absence of extracellular calcium (EGTA 1 mM). FC detected changes of (A) PLT Δψ and (B) formation of CD62p/PS-positive PLTs after ICU COVID-19 IgG incubation in vehicle or EGTA pretreated wPLTs, respectively. (C-D) ICU COVID-19 IgG-induced PLT changes in the presence (vehicle) or in intracellular calcium depleted (BAPTA 20 µM) wPLTs. FC detected changes of (C) PLT Δψ and (D) formation of CD62p/PS-positive PLTs induced by ICU COVID-19 IgG in vehicle or BAPTA preloaded wPLTs, respectively. Data are presented as mean percentage ± SEM of (A,C) Δψ depolarized PLTs and mean percentage ± SEM of (B,D) PS (Lactadherin-FITC) and CD62p-APC-positive wPLTs. Note that lactadherine is a calcium-independent marker of PS externalization. The number of patients and healthy donors tested is reported in each graph. See Figure 1 for P values and abbreviation definitions.
Similar effects were observed by depleting intracellular calcium stores. BAPTA pretreatment resulted in significant inhibition of ICU COVID-19 IgG-induced Δψ depolarization (percent Δψ depolarized PLTs ± SEM: 41.43 ± 3.80 vs 5.46 ± 1.17, P value = .0039; Figure 5C). In addition, BAPTA pretreatment of wPLTs resulted in marked prevention of ICU COVID-19 IgG-induced CD62p/PS-positive PLTs (percent CD62p/PS-positive PLTs: 29.58 ± 3.36 vs 1.74 ± 0.39, P value = .0020; Figure 5D; supplemental Figure 10).
Elevation of cAMP protects against COVID-19 IgG-induced procoagulant PLTs
The interplay between the signaling pathways of the intracellular second messengers, cAMP and calcium, has been shown to have an important role in numerous essential physiological processes during PLT activation and apoptosis.16,17 Therefore, we investigated the role of cAMP on COVID-19 antibody-induced formation of procoagulant PLTs. The pretreatment of PLTs with forskolin led to significant reduction of ICU COVID-19 IgG-induced Δψ depolarization (percent Δψ depolarized PLTs ± SEM: 27.80 ± 4.10 vs 6.60 ± 0.81, P value = .0079; Figure 6A). In addition, Forskolin pretreatment of wPLTs led to reduction of ICU COVID-19 IgG-induced procoagulant PLT generation (percent CD62p/PS-positive PLTs ± SEM: 35.29 ± 1.58 vs 12.26 ± 2.74, P value = .0313; Figure 6B; supplemental Figure 11). These findings indicate that the elevation of intracellular cAMP prevents ICU COVID-19 IgG-induced formation of procoagulant PLTs.

cAMP elevation prevents ICU COVID-19 IgG-induced formation of procoagulant PLTs. Panels (A-B) show ICU COVID-19 IgG-induced changes of procoagulant PLT markers in vehicle or Forskolin (2.25 µM) preincubated wPLTs. (A-B) ICU COVID-19 IgG-induced changes of (A) Δψ and (B) formation of CD62p/PS-positive PLTs were analyzed in vehicle or Forskolin preincubated wPLTs by FC. Panels (C-D) show ICU COVID-19 IgG-induced PLT changes in the presence of vehicle or Iloprost (20 nM). FC detected changes of (C) Δψ and (D) formation of CD62p/PS-positive PLTs induced by ICU COVID-19 IgG in vehicle or Iloprost preincubated wPLTs, respectively. (A,C) Data are presented as mean percentage ± SEM of Δψ depolarized PLTs and (B,D) mean percentage ± SEM of PS and CD62p-APC-positive-labeled wPLTs. The number of patients and healthy donors tested is reported in each graph. See Figure 1 for P values and abbreviation definitions.
cAMP elevation prevents ICU COVID-19 IgG-induced formation of procoagulant PLTs. Panels (A-B) show ICU COVID-19 IgG-induced changes of procoagulant PLT markers in vehicle or Forskolin (2.25 µM) preincubated wPLTs. (A-B) ICU COVID-19 IgG-induced changes of (A) Δψ and (B) formation of CD62p/PS-positive PLTs were analyzed in vehicle or Forskolin preincubated wPLTs by FC. Panels (C-D) show ICU COVID-19 IgG-induced PLT changes in the presence of vehicle or Iloprost (20 nM). FC detected changes of (C) Δψ and (D) formation of CD62p/PS-positive PLTs induced by ICU COVID-19 IgG in vehicle or Iloprost preincubated wPLTs, respectively. (A,C) Data are presented as mean percentage ± SEM of Δψ depolarized PLTs and (B,D) mean percentage ± SEM of PS and CD62p-APC-positive-labeled wPLTs. The number of patients and healthy donors tested is reported in each graph. See Figure 1 for P values and abbreviation definitions.
More importantly and of high clinical interest, a similar protective effect was observed with Iloprost, a clinically approved cAMP inducer.18 In fact, Iloprost pretreatment of wPLTs led to marked reductions of ICU COVID-19 IgG-induced Δψ depolarization (percent Δψ depolarized PLTs ± SEM: 37.14 ± 4.67 vs 13.29 ± 2.60, P value = .0078; Figure 6C). Remarkably, Iloprost (20 nM) pretreatment lead to a significant reduction of procoagulant CD62p/PS-positive PLTs (percent CD62p/PS-positive PLTs ± SEM: 41.36 ± 3.60 vs 22.22 ± 3.92, P value = .0156; Figure 6D; supplemental Figure 12). Noteworthy, no significant changes were observed in the number of CD62p single-positive PLTs (percent CD62p-positive PLTs ± SEM: 24.48 ± 2.2 vs 26.51 ± 3.94, P value = .6875; supplemental Figure 12).
Based on these findings, we were interested in whether Iloprost might affect the ability to form thrombus. Pretreatment of PLTs with Iloprost showed a marked reduction in ICU COVID-19 IgG-induced thrombus formation compared with vehicle (mean percent SAC ± SEM: 14.63 ± 2.31 vs 3.85 ± 0.95, respectively, P value = .0079; Figure 7A-B; supplemental Figure 13). Taken together, our data provide the first evidence for a potential therapeutic use of Iloprostin the treatment of the antibody-induced coagulopathy that is observed in COVID-19 disease.
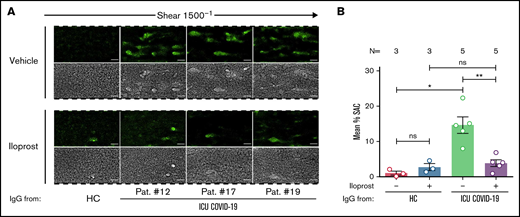
Iloprost inhibits ICU COVID-19 IgG-induced increased thrombus formation. (A) PRP from healthy individuals with the blood group O was incubated with HC or ICU COVID-19 IgG in the presence of vehicle or Iloprost (20 nM). After reconstitution into autologous whole blood, samples were perfused through microfluidic channels at a shear rate of 1500-1 (60 dyne) for 5 minutes. Pictures were acquired at x20 magnification in fluorescent (upper panel) as well as in the BF channel (lower panel). Scale bar 50 µm. (B) Mean percent of SAC by thrombus ± SEM induced by HC or ICU COVID-19 IgG in the presence of vehicle or Iloprost (20 nM). The number of patients and healthy donors tested is reported in each graph. See Figure 1 for P values and abbreviation definitions.
Iloprost inhibits ICU COVID-19 IgG-induced increased thrombus formation. (A) PRP from healthy individuals with the blood group O was incubated with HC or ICU COVID-19 IgG in the presence of vehicle or Iloprost (20 nM). After reconstitution into autologous whole blood, samples were perfused through microfluidic channels at a shear rate of 1500-1 (60 dyne) for 5 minutes. Pictures were acquired at x20 magnification in fluorescent (upper panel) as well as in the BF channel (lower panel). Scale bar 50 µm. (B) Mean percent of SAC by thrombus ± SEM induced by HC or ICU COVID-19 IgG in the presence of vehicle or Iloprost (20 nM). The number of patients and healthy donors tested is reported in each graph. See Figure 1 for P values and abbreviation definitions.
Discussion
Our study shows that IgGs from patients with severe COVID-19 are able to induce procoagulant PLTs with increased potential for thrombus formation via crosslinking FcγRIIA in a calcium-dependent manner. Most importantly, we observed that cAMP elevation by an approved drug, namely Iloprost, can sufficiently inhibit the initiation of procoagulant PLTs and the subsequent increase of thrombus formation by COVID-19 IgG antibodies. These data may have significant clinical implications.
It is well established that COVID-19 infection is associated with a systemic prothrombotic state and increased incidence of thromboembolic complications.19 However, the presequelae events leading to the hypercoagulopathy observed in COVID-19 still remain elusive. Data from our study demonstrate the presence of PLT-reactive IgG antibodies that harbor the ability to induce marked changes in PLTs in terms of increased Δψ depolarization, PS externalization, P-selectin expression, and GPVI shedding that are characteristic for procoagulant PLTs. A novel finding, and potentially of high clinical impact, is that antibody-mediated formation of procoagulant PLTs was associated with the clinical course as these changes progressively increased as patients needed ventilation and peaked between day 3 and day 7 in the ICU. The kinetics of these markers might have predictive value to monitor COVID-19-induced coagulopathy. In fact, PS externalization was recently found to be a predictive biomarker for thromboembolic complications related to cardiovascular therapeutic devices.20 In contrast to PS, conventional markers of PLT activation were declared in this study to fail the detection of early activation events leading to thrombosis. Of note, ICU COVID-19 IgG did not induce a single positive PLT population that expressed CD62P, indicating that PS externalization accounts for the enhance in double-positive PLTs. These data suggest activation of procoagulant pathways leading to PS externalization in addition to α-granule translocation.
The alterations in PLTs that were observed in our study after incubation with sera from patients with severe COVID-19 infection, such as Δψ disruption, BCL-XL reduction, and PS externalization, could be found in apoptotic as well as procoagulant PLTs. The involvement of PLT apoptosis to promote prothrombotic conditions has been controversially discussed. Recent data suggest that apoptotic PLTs are unable to promote prothrombotic conditions.10 Additionally, apoptotic PLTs have been described to show only a weak activation and aggregatory response.21 As our study showed that antibody-mediated Δψ disruption is associated with the induction of a CD62p/PS-positive PLT population, we suggest that IgG antibodies in COVID-19 induce procoagulant rather than apoptotic PLTs. This assumption is underlined as IgGs from COVID-19 patients also induced the cleavage of GPVI from the PLT surface that further indicates a procoagulant phenotype.22 Of note, 5 patients in our cohort received argatroban from the beginning of their ICU stay. Although we cannot exclude the presence of a minimal concentration of thrombin inhibitors in serum samples used in these experiments, the fact that changes in PLT phenotype were observed under heparin, as well as argatroban treatments, indicates rather an antibody-mediated than an anticoagulation effect. In fact, activated PLTs were shown elsewhere to be predominant in COVID-19 patients.6 In particular, CD62p-positive PLTs were suggested to be involved in thrombosis in COVID-19 via the interaction with neutrophil granulocytes leading to NET formation and increased interaction with the inflamed vessel wall. Here we show that COVID-19 IgG-antibodies have the capability to solely trigger a PLT population with dramatic procoagulant potential and that the interplay with other immune cells seems not to be a prerequisite. This hypothesis was supported by the findings of our in vitro studies where COVID-19-specific IgGs were able to trigger procoagulant PLT changes together with increased endogenous thrombin generation potential. Our findings were reinforced by data from an ex vivo microfluidic circulation system that revealed an increased thrombus formation in the presence of COVID-19 IgG antibodies. Contrary to these observations, recent data showed decreased stimulated procoagulant PLT responses in agonist-stimulated PLTs from COVID-19 patients.23 A potential explanation for this apparent discrepancy could be a different patient cohort or timing of blood collection. Another potential explanation, although speculative, could be that PLTs have lost their potential to produce a procoagulant phenotype due to permanent stimulation in COVID-19 that results in an exhausted PLT phenotype.
Motivated by these novel functional data, we thought to investigate the mechanistic pathways involved in the COVID-19 IgG-induced procoagulant status. Our data show that the formation of procoagulant PLTs is induced through the crosslinking of FcγRIIA by the Fc domain of IgG antibodies. Involvement of Fcγ receptors in severe COVID-19 infections were recently described by Combes et al.24 The blockade of FcγRIIA significantly inhibited ICU COVID-19 IgG-induced changes in Δψ depolarization. Most importantly, FcγRIIA blockade reduced procoagulant CD62p/PS-positive PLTs and subsequently inhibited COVID-19 antibody-induced ex vivo thrombus formation. These findings might suggest a transient onset of misdirected autoimmune mechanisms that result in the emergence of PLT-reactive antibodies in severe COVID-19 infection. We believe that until now, no further specified COVID-19 IgG-induced FcγRIIA-mediated signaling pathways trigger a distinct procoagulant PLT population that lead to increased thrombus formation. While these findings are novel for COVID-19-associated hypercoagulopathy, the correlation between FcγRIIA engagement and increased risk for thromboembolic events is well established for heparin-induced thrombocytopenia (HIT).25 Interestingly, the IgG antibody formation peaks in HIT within 5 to 10 days after exposure to heparin and is associated with PLT consumption and increased risk for thrombosis.26,27 Typical HIT antibodies were not detected in sera that induced procoagulant PLTs in our study (data not shown). However, in our study, the ability of ICU COVID-19 sera to induce procoagulant PLTs was most pronounced between day 3 and day 7. The glycosylation profile of IgG antibodies has been recently shown to change during COVID-19 infection.28 Our data may indicate that the interplay between IgG and PLTs is modulated by multifactorial components. The variation that is observed in our study in the ability of patients’ sera to induce a procoagulant PLT phenotype might be explained by temporal changes in the glycosylation pattern of patients’ IgG (ie, afucosylation). On the other hand, polymorphisms in FcγRIIA could be one explanation for the interindividual variation in PLT response to COVID-19 antibodies.
Another minor finding in our study is the greater disruption of Δψ compared with PS externalization. This might be due to the timing of sampling and differences in the methodology. Further investigations are required in the future to characterize PLTs that have lost mitochondrial membrane potential but still externalize low levels of PS.
Calcium is involved in many biological mechanistic pathways in PLTs.16 In response to PLT agonists, calcium is released from the PLT internal stores, which is followed by amplifying second wave extracellular calcium influx via store (SOCE) and receptor-operated calcium entry (ROCE), respectively.29 The role of calcium signaling in ITAM (immunoreceptor tyrosine-based activation motif) coupled with GPVI and (hem)ITAM coupled CLEC-2 receptor signaling has been well established in the last few years.30 Additionally, FcγRIIA ligation has been shown to induce calcium mobilization via ITAM signaling prior to PLT aggregation.31 In our study, the depletion of calcium in the extracellular compartment inhibited ICU COVID-19 IgG-induced procoagulant changes. These findings confirm previous studies that demonstrated the pivotal role of calcium signaling in the formation of procoagulant PLTs.9,32,33 Three calcium-signaling events were proposed to be involved in this process, (1) agonist-induced events leading to (2) an increase in cytosolic calcium and subsequent mitochondrial signal that finally results in (3) supramaximal cytosolic calcium signal.34 It is currently unclear whether this threshold concept also applies to the procoagulant PLT phenotype that occurs via crosslinking the FcγRIIA by IgG from COVID-19.
Another possible explanation for the inhibition of COVID-19 antibody-induced procoagulant PLTs in the absence of extracellular calcium could be the loss of distinct calcium-dependent conformational properties of PLT epitopes that are targeted by COVID-19 IgG antibodies. Dimeric PLT GPIIb/IIIa, the receptor for fibrinogen and well-known immunogenic epitope of PLT-reactive autoantibodies in immune thrombocytopenia (ITP), has been well characterized to be structurally dependent on extracellular calcium levels.35 Although this might be currently too speculative, the depletion of extracellular calcium might have inhibited antibody binding to such conformation-sensitive epitopes as GPIIb/IIIa. ICU COVID-19 IgG-induced PLT alterations were dependent on extracellular as well as intracellular calcium levels. Since PLT apoptosis has been described to be independent of calcium,36 our finding indicates that COVID-19 IgGs trigger FcγRIIA-mediated events that result in procoagulant PLT formation in a calcium-dependent manner. Future studies could explore the exact mechanisms by which calcium contributes to procoagulant PLT formation in COVID-19.
COVID-19 antibody-induced procoagulant PLTs were significantly inhibited by the use of inducers of adenylate cyclase (AC) that is well known to cause increased cAMP levels in PLTs.37 The protective effect of cAMP was demonstrated, as Iloprost, an already approved prostacyclin derivate and inducer of AC, efficiently prevented the formation of procoagulant PLTs in response to COVID-19 antibodies. Iloprost use is also described to be safe in patients undergoing cardiac surgery with diagnosed FcγRIIA-dependent HIT or with HIT-reactive antibodies.38 Finally, and of high clinical importance, Iloprost pretreatment of PLTs markedly reduced COVID-19 IgG-induced thrombus formation on collagen, suggesting that cAMP inducers may have the potential to prevent life-threatening thromboembolic events in COVID-19 antibody-mediated coagulopathy. Another minor finding from our microfluidic system was that Iloprost, despite significantly inhibited antibody-mediated thrombus formation, did not affect the CD62p-single-positive PLT population. Since Iloprost prevented thrombus formation, this might indicate that PS rather than CD62p exposure on the PLT surface is pivotal to trigger the onset of thromboembolic events. Although Iloprost showed a significant inhibition of the antibody-induced FcγRIIA-mediated generation of procoagulant PLTs, additional mechanistic studies are still needed to clarify whether the agent specifically impacts the FcγRIIA-initiated procoagulant PLT formation or acts as a general inhibitor of PLT responses.
Our study is subjected to some limitations. First, as an observational, monocentric study, we cannot conclude that the reported associations between IgG antibodies and changes in markers for procoagulant PLTs in COVID-19 are causal for the thrombosis or specific for the disease. Second, we cannot exclude the possibility of remaining residual confounding or unmeasured potential confounders in our mechanistic studies. Third, the low number of patients does not enable a final and robust statistical analysis to assess clinical outcomes in patients with increased procoagulant PLTs. Finally, although our live-imaging studies indicate multicellular thrombus formation, increased PLT accumulation or adherence in our ex vivo model cannot be excluded. Nevertheless, data presented in this study may provide a background for future studies to dissect mechanisms related to PLT activation that are involved in the progression of COVID-19.
Taken together, our study shows that IgG antibodies from patients with severe COVID-19 are able to stimulate FcγRIIA, leading to the induction of procoagulant PLTs with an increased ability of thrombus formation. These processes are dependent on calcium and can be efficiently inhibited by cAMP inducers, suggesting that AC might represent a potentially promising target to prevent thromboembolic complications in COVID-19 disease.
Acknowledgments
The authors thank Juliane Wolf for her excellent technical support and Susanne Staub for editing the article as a native English speaker.
This work was supported by grants from the Ministerium für Wissenschaft, Forschung und Kunst Baden-Württemberg (J.Z. and T.B.), German Red Cross and the Herzstiftung (T.B.) (BA5158/4 and TSG-Study), and TÜFF-Gleichstellungsförderung (K.A.) (2563-0-0). This project was supported by the German Research Foundation (DFG), project number 374031971 – TRR 240.
Authorship
Contribution: J.Z., K.A., T.B., and P.R. designed the study; P.R. and H.H. were responsible for the treatment of the patients; K.A. and H.H. collected and analyzed the clinical data; J.Z., K.A., H.J., L.P., A.S., A.W., and K.W. performed the experiments; J.Z., K.A., H.J., L.P., A.S., and A.W. collected the data; J.Z., K.A., H.J., L.P., A.S., A.W., M.B., N.M., S.G., H.B., M.G., V.M., P.R., and T.B. analyzed the data, interpreted the results, and wrote the manuscript; and all authors read and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Tamam Bakchoul, University Hospital of Tuebingen, Otfried-Müller-Straße 4/1, 72076 Tübingen, Germany; e-mail: tamam.bakchoul@med.uni-tuebingen.de.

