

Key Points
TCRαβ/CD19-depleted HLA-haploidentical HSCT is an effective strategy for children with several nonmalignant disorders.
Patients given this type of transplant benefit from a low incidence of GVHD and TRM, with graft failure being the main obstacle.

Abstract
Several nonmalignant disorders (NMDs), either inherited or acquired, can be cured by allogeneic hematopoietic stem cell transplantation (HSCT). Between January 2012 and April 2020, 70 consecutive children affected by primary immunodeficiencies, inherited/acquired bone marrow failure syndromes, red blood cell disorders, or metabolic diseases, lacking a fully matched donor or requiring urgent transplantation underwent TCRαβ/CD19-depleted haploidentical HSCT from an HLA-partially matched relative as part of a prospective study. The median age at transplant was 3.5 years (range 0.3-16.1); the median time from diagnosis to transplant was 10.5 months (2.7 for SCID patients). Primary engraftment was obtained in 51 patients, while 19 and 2 patients experienced either primary or secondary graft failure (GF), the overall incidence of this complication being 30.4%. Most GFs were observed in children with disease at risk for this complication (eg, aplastic anemia, thalassemia). All but 5 patients experiencing GF were successfully retransplanted. Six patients died of infectious complications (4 had active/recent infections at the time of HSCT), the cumulative incidence of transplant-related mortality (TRM) being 8.5%. Cumulative incidence of grade 1-2 acute GVHD was 14.4% (no patient developed grade 3-4 acute GVHD). Only one patient at risk developed mild chronic GVHD. With a median follow-up of 3.5 years, the 5-year probability of overall and disease-free survival was 91.4% and 86.8%, respectively. In conclusion, TCRαβ/CD19-depleted haploidentical HSCT from an HLA-partially matched relative is confirmed to be an effective treatment of children with NMDs. Prompt donor availability, low incidence of GVHD, and TRM make this strategy an attractive option in NMDs patients. The study is registered at ClinicalTrial.gov as NCT01810120.
Introduction
Indications to allogeneic hematopoietic stem cell transplantation (HSCT) in children comprise a wide range of different nonmalignant disorders (NMDs), either congenital (eg, thalassemia) or acquired such as severe aplastic anemia.1 Every year, new genetic disorders, many of which involving innate and/or adaptive immunity, are identified and characterized, thus widening the number of diseases that can benefit from HSCT.2 In recent years, gene therapy, either as gene addition3 or genome-editing,4 is becoming a clinical reality. However, since specific vectors or genome-editing targets must be identified, developed, and studied individually for each disease-causing gene, HSCT remains the sole curative option for several different conditions.
Historically, for treating NMDs, the best results have been achieved using an HLA-identical sibling. However, the probability of identifying a fully compatible, nonaffected sibling is theoretically less than 25%; in western countries, it has been calculated that, mainly because of low birth rate, this probability drops to less than 20% in the first 5 years of life.5 On the other side, the probability of finding a fully matched unrelated donor (MUD) depends mainly on the ethnicity of the patient, this reflecting the number of donors present in the international databases.6 White patients of European descent have the highest probability of finding such a donor (around 75%), while Blacks of South or Central American descent have the lowest (16%). Moreover, unmanipulated HSCTs from an unrelated donor carry an increased risk of developing both acute and chronic graft-versus-host disease (GVHD), complications that are particularly detrimental in children with NMDs.
In light of these considerations, strategies to exploit alternative donors have been developed. Among them, improvement in the techniques of ex-vivo T cell depletion,7 a type of graft manipulation coupled with the use of mega-doses of stem cells,8 made the use of haploidentical donors safer, reducing the risk of GVHD.
We previously reported our pivotal experience with the selective depletion of TCRαβ+ lymphocytes (ie, the lymphocyte subset mainly responsible for GVHD), coupled with the depletion of CD19+ B cells (performed with the goal of preventing the occurrence of EBV-driven posttransplant lymphoproliferative disorders [PTLD]), in 23 children affected by different NMD.9 Incidence of acute GVHD was low, while we did not observe cases of chronic GVHD; overall survival (OS) and disease-free survival (DFS) resulted above 90%. This experience was subsequently paralleled by other groups which focused mainly on primary immunodeficiencies (PIDs).10-13 All these studies confirmed the low incidence of both acute and chronic GVHD, with encouraging survival results, infections being the main cause of transplant-related mortality (TRM).
Here we report on the outcomes of 70 patients affected by different NMDs who received a TCRαβ+/CD19+-depleted haplo-HSCT in the context of a prospective, single-center phase 2 trial (#NCT01810120).
Methods
Patients and transplant
Children (age < 21 years) with NMDs deemed curable with an allogeneic HSCT and lacking a related or 10/10 allelic-matched unrelated donor (or in need of an urgent procedure [ie, within 2 months from recognition of the indication] according to physician’s judgment) and treated between 01/2012 and 04/2020 were enrolled in a single-center prospective clinical trial (registered at ClinicalTrial.gov #NCT01810120). The study was approved by the Ethical Committee of the Ospedale Pediatrico Bambino Gesù. Written informed consent was obtained from the patient’s parents or legal guardian. The trial was conducted in accordance with the Helsinki Declaration.
Details on patient/donor characteristics are reported in Table 1. All patients received a pretransplant conditioning regimen that differed according to the original disorder (Table 1). Anti-T lymphocyte globulin (ATLG Grafalon, Neovii Biotech) was administered at a dose of 12 mg/kg over 3 days (ie, from day −5 to −3). On day −1, patients received rituximab (200 mg/m2) for in vivo depletion of both donor and recipient B cells. No patient was given any posttransplantation pharmacologic GVHD prophylaxis.
Patient, donor, and transplant characteristics
| Patients | N (%) | Notes |
| Sex | ||
| Male | 43 (61) | |
| Female | 27 (39) | |
| Age at diagnosis, median (range), y | 0.96 (0.0 (prenatal)-14.2) | |
| Age at HSCT, median (range), y | 3.5 (0.3-16.1) | |
| Time from diagnosis to HSCT, median (range), mo | 10.5 (1.2-177.7) | |
| for SCID patients, mo | 2.7 (1.3-16.8) | |
| Diagnosis | n (%) | |
| SCID | 21 [3 Omenn] (30) | RAG1-2: 8; IL2RG: 3; JAK3: 2; ADA: 2; PNP: 1; DCLRE1C: 1; CD3e: 1; IL7R: 1; unk: 2 |
| SAA* | 13 (18.5) | |
| RBC disorders | 11 (15.5) | Thal: 10; SCD: 1 |
| HLH | 6 (8.5) | |
| Other PID | 11 (15.5) | CID: 2 (1 IL7R; 1 RAG1-2); WAS: 2; IPEX: 1; HIES: 1; CGD: 1; CD27def: 1; WHIM: 1; LRBA-def: 1; NEMO-def: 1 |
| IBMFS | 3 (4.5) | CAMT: 1; SDS: 1; SCN: 1 |
| Metabolic disorders | 3 (4.5) | ALD: 2; MLD: 1 |
| Other | 2 (3) | Osteopetrosis: 1; NMO: 1 |
| Previous HSCT | 5 (7) | |
| Donor characteristics | median (range) | |
| Age, y | 37.5 (20-51) | |
| Type of donor | n (%) | |
| Mother | 41 (58.5) | |
| Father | 26 (37) | |
| Sibling | 3 (4.5) | |
| Sex mismatch | 35 (50) | |
| Female donor → male recipient | 26/35 (74) | |
| HLA-match | ||
| Host-versus-graft direction | n (%) | |
| 5/10 | 42 (60) | |
| 6/10 | 20 (28.5) | |
| 7/10 | 8 (11.5) | |
| Graft-versus-host direction | n (%) | |
| 5/10 | 32 (46) | |
| 6/10 | 22 (31.5) | |
| 7/10 | 16 (22.5) | |
| CMV status for donor/recipient | n (%) | |
| NEG/NEG | 3 (4.5) | |
| POS/NEG | 10 (14) | |
| NEG/POS | 7 (10) | |
| POS/POS | 50 (71.5) | |
| Infectious status at HSCT | n (%) | |
| No infections | 47 (67) | |
| Infection present | 23 (33) | |
| Bacterial | 3 (13) | |
| Viral | 17 (74) | |
| Fungal | 3 (13) | |
| Conditioning regimen used | n (%) | |
| Busulfan†+Thiotepa+Fludarabine | 24 (34) | Thal, SCD, HLH, metabolic disorders, some PIDs |
| Treosulfan+Thiotepa+Fludarabine | 18 (26) | some PIDs, IBMFs, other |
| Treosulfan+Fludarabine | 16 (23) | SCID |
| Cyclophosphamide+Fludarabine ±TBI | 11 (15.5) | SAA |
| Other | 1 (1.5) | |
| Cell dose infused | median (range) | |
| CD34+ cells × 106/kg | 20.3 (8.5-48.8) | |
| αβ+ T cells × 106/kg | 0.034 (0.002-0.095) | |
| γδ+ T cells × 106/kg | 13.0 (1.0-143.4) | |
| NK cells × 106/kg | 48.5 (8.1-156.1) | |
| CD20+ cells × 106/kg | 0.02 (0.003-1.96) |
| Patients | N (%) | Notes |
| Sex | ||
| Male | 43 (61) | |
| Female | 27 (39) | |
| Age at diagnosis, median (range), y | 0.96 (0.0 (prenatal)-14.2) | |
| Age at HSCT, median (range), y | 3.5 (0.3-16.1) | |
| Time from diagnosis to HSCT, median (range), mo | 10.5 (1.2-177.7) | |
| for SCID patients, mo | 2.7 (1.3-16.8) | |
| Diagnosis | n (%) | |
| SCID | 21 [3 Omenn] (30) | RAG1-2: 8; IL2RG: 3; JAK3: 2; ADA: 2; PNP: 1; DCLRE1C: 1; CD3e: 1; IL7R: 1; unk: 2 |
| SAA* | 13 (18.5) | |
| RBC disorders | 11 (15.5) | Thal: 10; SCD: 1 |
| HLH | 6 (8.5) | |
| Other PID | 11 (15.5) | CID: 2 (1 IL7R; 1 RAG1-2); WAS: 2; IPEX: 1; HIES: 1; CGD: 1; CD27def: 1; WHIM: 1; LRBA-def: 1; NEMO-def: 1 |
| IBMFS | 3 (4.5) | CAMT: 1; SDS: 1; SCN: 1 |
| Metabolic disorders | 3 (4.5) | ALD: 2; MLD: 1 |
| Other | 2 (3) | Osteopetrosis: 1; NMO: 1 |
| Previous HSCT | 5 (7) | |
| Donor characteristics | median (range) | |
| Age, y | 37.5 (20-51) | |
| Type of donor | n (%) | |
| Mother | 41 (58.5) | |
| Father | 26 (37) | |
| Sibling | 3 (4.5) | |
| Sex mismatch | 35 (50) | |
| Female donor → male recipient | 26/35 (74) | |
| HLA-match | ||
| Host-versus-graft direction | n (%) | |
| 5/10 | 42 (60) | |
| 6/10 | 20 (28.5) | |
| 7/10 | 8 (11.5) | |
| Graft-versus-host direction | n (%) | |
| 5/10 | 32 (46) | |
| 6/10 | 22 (31.5) | |
| 7/10 | 16 (22.5) | |
| CMV status for donor/recipient | n (%) | |
| NEG/NEG | 3 (4.5) | |
| POS/NEG | 10 (14) | |
| NEG/POS | 7 (10) | |
| POS/POS | 50 (71.5) | |
| Infectious status at HSCT | n (%) | |
| No infections | 47 (67) | |
| Infection present | 23 (33) | |
| Bacterial | 3 (13) | |
| Viral | 17 (74) | |
| Fungal | 3 (13) | |
| Conditioning regimen used | n (%) | |
| Busulfan†+Thiotepa+Fludarabine | 24 (34) | Thal, SCD, HLH, metabolic disorders, some PIDs |
| Treosulfan+Thiotepa+Fludarabine | 18 (26) | some PIDs, IBMFs, other |
| Treosulfan+Fludarabine | 16 (23) | SCID |
| Cyclophosphamide+Fludarabine ±TBI | 11 (15.5) | SAA |
| Other | 1 (1.5) | |
| Cell dose infused | median (range) | |
| CD34+ cells × 106/kg | 20.3 (8.5-48.8) | |
| αβ+ T cells × 106/kg | 0.034 (0.002-0.095) | |
| γδ+ T cells × 106/kg | 13.0 (1.0-143.4) | |
| NK cells × 106/kg | 48.5 (8.1-156.1) | |
| CD20+ cells × 106/kg | 0.02 (0.003-1.96) |
ALD, adrenoleukodystrophy; CAMT, congenital amegakaryocytic thrombocytopenia; FA, Fanconi anemia; IBMFS, inherited bone marrow failure syndromes; mo, months; NEMO, Nuclear factor-kappa B essential modulator (NEMO) deficiency; SAA, severe aplastic anemia; unk, unknown; WAS, Wiskott-Aldrich syndrome; y, years.
1 patient with EB and SAA
busulfan was adjusted to maintain Css between 600 and 900 ng/ml
Donor selection criteria, as well as donor mobilization and graft manipulation, are detailed in supplemental data.9,14,15 Chimerism analysis was performed weekly for the first 3 months and monthly thereafter. Immune recovery (absolute number of CD3+, TCRαβ+, TCRγδ+, CD4+, CD8+, NK, and CD19+ cells) was evaluated at 1, 3, 6, 12, and 18 months after transplantation.
Statistical analysis
Details on statistical analysis are reported in supplemental data. Survival outcomes, namely OS, event-free survival (EFS) and DFS (defined as the probability of survival, without evidence of disease at time of last follow-up [ie, patients receiving a successful second allograft after GF and without recurrence of the disease at last follow-up were considered disease-free]) were calculated using the Kaplan-Meier method. TRM was defined as the probability of death from any transplant-related cause. We also evaluated the composite end-point of chronic GVHD-free and relapse-free survival (GRFS) as defined by Holtan et al.16
Acute and chronic GVHD were diagnosed and graded according to previously published criteria.17,18 Acute and chronic GVHD, TRM, and graft failure (GF) were expressed as cumulative incidences in order to adjust the estimations for competing risks (see supplemental data for details).19,20
Quantitative variables were reported as median value and range, while categorical variables were expressed as absolute value and percentage. Categorical variables were compared using the χ-square test or Fisher’s exact test, as appropriate, while the Mann-Whitney U rank-sum test or the Student t test were used for continuous variables. The significance of differences between OS, DFS, and GRFS was estimated by the log-rank test, while Gray's test was used to assess differences between cumulative incidences. Multivariate analysis was performed using the Cox proportional hazard regression model.
Viral infections were assessed as percentage, cumulative incidence, and time-averaged area-under-the-curve (AAUC).21,22
Statistical analysis was performed using EZR version 1.32 (Saitama Medical Centre, Jichi Medical University),23 a graphical user interface for R (The R Foundation, Vienna, Austria; http://www.R-project.org) and GraphPad version 8.0 (GraphPad Software, San Diego, CA).
Results
Engraftment/GF
Seventy children were enrolled in the study between January 2012 and April 2020. Of these, 20 patients have been previously reported.2,9 Details on patient, donor, and transplantation characteristics are reported in Table 1. Sixty-five patients received haplo-HSCT as a first allograft. The remaining 5 had failed a previous one (1 mismatched unrelated cord blood transplant [UCBT], 1 mismatched unrelated donor, 1 MUD, and 2 haploidentical HSCT using different platforms).
All patients but one (a 10-year-old girl affected by SAA) received more than 10x106 CD34+ cells/kg; in no case was the threshold of 1x105 TCRαβ+ cells/kg exceeded. Patients with hemophagocytic lymphohistiocytosis (HLH), thalassemia, SAA, or osteopetrosis were considered at high risk of developing GF,24-26 while patients affected by other NMDs were considered standard risk. Fifty-one patients (72.8%) achieved sustained primary donor cell engraftment; of them, 2 patients experienced secondary GF. The cumulative incidence of either primary (n = 19) or secondary (n = 2) GF was 30.4% (95% confidence interval [CI] 21.0-42.7) (Figure 1A). Median time to neutrophil and platelet recovery was 14.5 (range 9-33) and 10 days (range 7-51), respectively (supplemental Figure 1A). A lower number of CD34+ cells infused with the graft (ie, below the first quartile) was associated with delayed recovery of platelets, but not of neutrophils (supplemental Figure 1B). As expected, GF occurred more frequently in children with disorders known to be associated with an increased GF risk (see also Figure 1B). Of the 3 patients affected by metabolic disorders, one with metachromatic leukodystrophy experienced GF and was rescued with a second HSCT. Neither composition of the graft (ie, number of CD34+, TCRαβ+, TCRγδ+, NK cells infused) nor the number of HLA-mismatches in the host-versus-graft (HvG) direction correlated with the risk of GF (data not shown). Sex mismatch was associated with a trend toward reduced incidence of GF (supplemental Figure 2). This factor was significant (together with the type of disease) in multivariable analysis (supplemental Table 1). Sixteen of the 21 patients with either primary or secondary GF were successfully retransplanted (2 with a mismatched UCBT, the other having received a second TCRαβ/CD19-depleted haploHSCT from the other parent). The median time interval between the first and the second transplant and between rejection and the second transplant was 40 days (range 22-76) and 22 days (range 10-51), respectively. Supplemental Table 2 reports details on conditioning regimens used, graft characteristics, and complications occurring after the second HSCT.
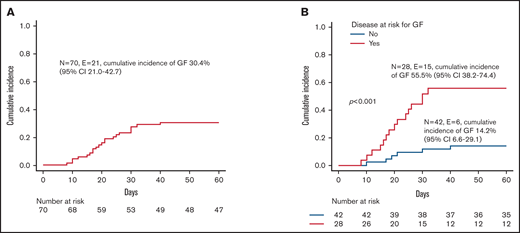
Graft failure. (A) Cumulative incidence of GF for the whole cohort. (B) Cumulative incidence of GF for patients affected by diseases at high risk (ie, children affected by HLH, thalassemia, SAA, or osteopetrosis; red line) or at standard risk (blue line) for the complication.
Graft failure. (A) Cumulative incidence of GF for the whole cohort. (B) Cumulative incidence of GF for patients affected by diseases at high risk (ie, children affected by HLH, thalassemia, SAA, or osteopetrosis; red line) or at standard risk (blue line) for the complication.
Three children (1 case each of HLH, SAA, and SCID) experiencing GF died because of infectious complications (1 case each of infection from multidrug resistant P aeruginosa, Vancomycin-resistant Enterococcus, and Pneumocystis jirovecii) before second transplantation; 2 additional patients (affected by thalassemia), failed a second transplant and were thus infused with back-up autologous stem cells. Thus, 4 out of the 5 patients who were not successfully retransplanted belong to the high-risk group. As already reported,27 GF was associated with HLH-like features, including high-grade fever, increased ferritin and LDH, hepato-/splenomegaly, hemophagocytosis in the BM. Neither high-dose steroids nor the use of calcineurin inhibitors were able to rescue these patients once that rejection had started.
One year after HSCT, 9 (15.5%) out of the 58 evaluable patients had a mixed chimerism (median of 76% donor cells on whole blood, range 40-96; supplemental Figure 3). Six were transplanted because of SCID (1 for Omenn), 2 for HLH, and 1 for NEMO deficiency. All received more than 20x106 CD34+ cells/kg with the graft. Six out of these 9 patients experienced viral infections in the early posttransplant period. None of the patients had complications related to the mixed chimerism; in particular, for the 2 HLH patients, it was 96% and 95%, respectively, far above the “critical” threshold of 20% to 30%.28
GVHD
Despite the fact that we did not administer any posttransplant pharmacological GVHD prophylaxis, incidence of both acute and chronic GVHD was low. In particular, 16 children developed skin-only, grade 1-2 acute GVHD (corresponding to a cumulative incidence of 24.2% [95% CI 15.0-34.8]) (Figure 2A); since 6 patients experienced grade 1 GVHD, the cumulative incidence of clinically relevant grade 2 GVHD was 14.4% (95% CI 7.4-23.7) (Figure 2B). Patients were successfully treated with low-dose steroids (ie, 0.5-1 mg/kg/die), calcineurin inhibitors, or extracorporeal photochemotherapy; the treatment was chosen mainly on patients’ age and infectious status. Neither graft composition nor other clinical variables had an influence on the occurrence of acute GVHD. We registered only 1 case of de novo mild chronic GVHD in a patient with SAA who, after experiencing a primary GF, was given a mismatched UCBT.
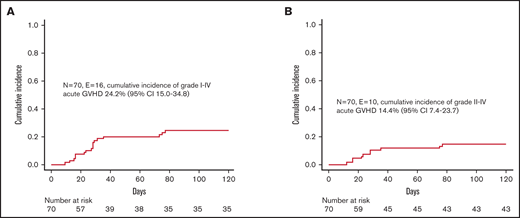
Graft-versus-host disease. (A) Cumulative incidence of grade 1-4 acute GVHD. (B) Cumulative incidence of grade 2-4 acute GVHD.
Graft-versus-host disease. (A) Cumulative incidence of grade 1-4 acute GVHD. (B) Cumulative incidence of grade 2-4 acute GVHD.
Infections and TRM
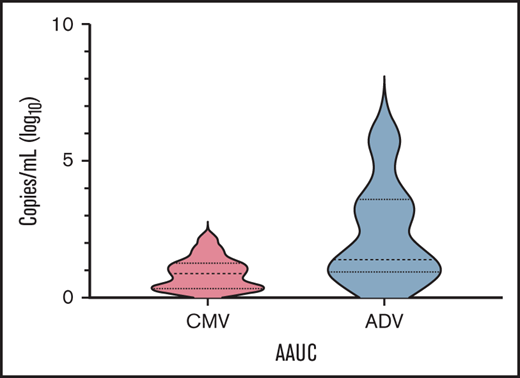
Table 2 reports details on infectious complications after transplant. The most frequent ones were CMV and adenovirus (ADV) infections, with a cumulative incidence of 27.7% (95% CI 18.6-39.9) and 10.1% (95% CI 4.9-20.0), respectively, followed by bacterial infections and viral respiratory infections. Since cumulative incidence is an inaccurate method of reporting posttransplant herpesvirus and ADV infections (eg, it does not take into account viral load and time of positivity/treatment), we calculated the AAUC180, as previously described.21,22 In details, median AAUC180 for CMV and ADV were 0.91 (range 0.21-1.96) and 1.42 (range 0.42-5.73) (Figure 4), respectively. No patient developed PTLD.
Posttransplant infections observed in the whole cohort
| Type of infection | n (%) |
|---|---|
| Viral | |
| CMV | 19 (27) |
| ADV | 7 (10) |
| HHV6 | 3 (4) |
| EBV | 2 (3) |
| BK virus (symptomatic) | 2 (3) |
| Respiratory* | 7 (10) |
| Gastrointestinal† | 3 (4) |
| Bacterial | |
| Pseudomonas aeruginosa | 5 (7) |
| Klebsiella pneumoniae | 5 (7) |
| CONS | 6 (8.5) |
| Enterococcus spp | 3 (4) |
| E coli | 1 (1.5) |
| C difficile | 1 (1.5) |
| Branhamella catarrhalis | 1 (1.5) |
| Fungal | |
| Candida spp | 2 (3) |
| Aspergillus spp | 1 (1.5) |
| Type of infection | n (%) |
|---|---|
| Viral | |
| CMV | 19 (27) |
| ADV | 7 (10) |
| HHV6 | 3 (4) |
| EBV | 2 (3) |
| BK virus (symptomatic) | 2 (3) |
| Respiratory* | 7 (10) |
| Gastrointestinal† | 3 (4) |
| Bacterial | |
| Pseudomonas aeruginosa | 5 (7) |
| Klebsiella pneumoniae | 5 (7) |
| CONS | 6 (8.5) |
| Enterococcus spp | 3 (4) |
| E coli | 1 (1.5) |
| C difficile | 1 (1.5) |
| Branhamella catarrhalis | 1 (1.5) |
| Fungal | |
| Candida spp | 2 (3) |
| Aspergillus spp | 1 (1.5) |
CONS, coagulase negative staphylococci.
4 rhinovirus (in 3 patients it was a coinfection with other respiratory viruses), 2 RSV, 1 each influenza B, parainfluenzae 3, non-SARS-CoV-2 coronavirus, bocavirus.
2 rotavirus, 1 enterovirus.
Six patients died of infectious complications at a median time of 85 days after HSCT (range 29-140), the 5-year cumulative incidence of TRM being 8.5% (95% CI 3.9-18.0) (Figure 3A). TRM of patients with an active or recent (ie, within the month preceding TCRαβ+/CD19-depleted haploHSCT) infection at the time of transplant was 17.3% (95% CI 6.9-39.9) as compared with a value of 4.2% (95% CI 1.0-16.0) (P = .06) for those without infections (supplemental Figure 4). As reported above, the 3 patients who died after GF were colonized by the bacteria, or, in the case of SCID patient, were affected by Pneumocistis jirovecii pneumonia before HSCT. The remaining 3 patients (affected by SAA, CAMT, and SCID) suffered from uncontrollable viral infections (2 cases of disseminated ADV infection and 1 case of CMV pneumonia). No case of GVHD-related death was recorded.

Survival. (A) Overall survival (OS) and transplant-related mortality (TRM) of the whole cohort. (B) Event-free survival of the whole cohort. (C) Disease-free survival of the whole cohort (excluding 2 patients with ALD, who had CNS involvement at the time of HSCT). (D) OS according to infectious status at transplant (ie, active or recent infection at the time of HSCT versus no infection).
Survival. (A) Overall survival (OS) and transplant-related mortality (TRM) of the whole cohort. (B) Event-free survival of the whole cohort. (C) Disease-free survival of the whole cohort (excluding 2 patients with ALD, who had CNS involvement at the time of HSCT). (D) OS according to infectious status at transplant (ie, active or recent infection at the time of HSCT versus no infection).
Survival
With a median follow-up of 3.7 years (range 1-9.3) for surviving patients, OS and EFS (considering both primary and secondary GF as events) are 91.4% (95% CI 81.9-96.1) and 65.7% (95% CI 53.4-75.5), respectively (Figure 3A-B). Given that most events were represented by GF, factors influencing EFS were represented by those associated with GF. Since most patients experiencing GF were successfully retransplanted, DFS is 86.8% (95% CI 76.1-92.9) (Figure 3C) (2 patients, one each affected by adrenoleukodystrophy and metachromatic leukodystrophy, who were transplanted because of rapid worsening of neurological symptoms, were excluded from this analysis). For the same reason, the occurrence of GF did not affect the probability of OS (93.9% for non-GF patients compared with 85.7% of GF patients; P = .3). Similarly, a previous failed HSCT did not significantly affect survival outcomes (data not shown). Only an active/recent infection influenced OS; indeed, OS of patients with active or recent infection at HSCT was 82.6% (95% CI 60.1-93.1) as compared with 95.7% (95% CI 84.0-98.9) of those without infections (P = .056) (Figure 3D). Notably, no donor/graft composition characteristic was associated with the outcome. Regarding patients affected by inborn errors of immunity (N = 38), OS was 92.1% (95% CI 77.5-97.4). More in details, OS for SCID patients (n = 21) and non-SCID PID patients (n = 17) was 90.5% (95% CI 67.0-97.5) and 94.1% (95% CI 65.0-99.1), respectively (p = n.s.; supplemental Figure 5).
Because of the low incidence of both acute and chronic GVHD, GRFS for the whole cohort was 87.1% (95% CI 76.7-93.1) and was not influenced by patients, donors, or transplant’s characteristics.
Immune reconstitution
Figure 5 reports details on immune reconstitution of the study population. As previously described in this transplant setting,9,29 the early posttransplant period is characterized by prompt recovery of TCRγδ+ and NK cells, followed by the progressive emergence of TCRαβ+ lymphocytes. Although CD3+ cell recovery was fast, CD3+CD4+ cell counts approached 500/μl only at 6 months after HSCT. Of the 64 patients alive, 58 (90.5%) were independent from immunoglobulin (Ig) replacement therapy at last follow-up. Notably, 4 out of 6 Ig-dependent patients showed a mixed chimerism, while 2 had experienced acute GVHD.
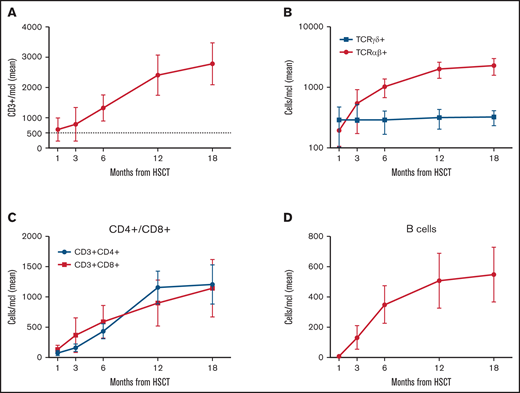
Immune reconstitution. (A) Absolute number after haplo-HSCT of CD3+ cells (mean and 95% CI). (B) TCRαβ+ and TCRγδ+ cells (mean and 95% CI). (C) CD4+ and CD8+ T cell subsets (mean and 95% CI). (D) CD20+ B cells (mean and 95% CI).
Immune reconstitution. (A) Absolute number after haplo-HSCT of CD3+ cells (mean and 95% CI). (B) TCRαβ+ and TCRγδ+ cells (mean and 95% CI). (C) CD4+ and CD8+ T cell subsets (mean and 95% CI). (D) CD20+ B cells (mean and 95% CI).
Immune-mediated complications
Five patients (2 SCID, 1 CID, 1 CD27 deficiency, and 1 warts-hypogammaglobulinemia, infections and myelokathexis syndrome, WHIM) developed autoimmune complications: 2 isolated autoimmune-hemolytic anemia (AIHA), 1 AIHA and Guillain-Barrè syndrome (GBS), 1 immune-mediate thrombocytopenia (ITP) and 1 autoimmune neutropenia (AIN). The patient with AIN already had this complication before the HSCT. Four patients had full donor chimerism on peripheral blood, while the last, with AIHA, had mixed chimerism (80% donor cells on peripheral blood). One patient had resolution of AIHA after steroids, while the patient with ITP fully recovered after rituximab treatment; the 3 others obtained a complete response after multiple lines of immune-suppressive therapy.
Discussion
Here we report the outcome of a cohort of 70 pediatric patients affected by several different NMDs, ranging from inborn errors of immunity to metabolic disorders, with many patients having a follow-up longer than 5 years. Nineteen patients were already reported in 20149 ; none of them experienced long-term events. Moreover, despite broadening the variety of diseases transplanted (which, however, have peculiar transplant-related issues), OS and DFS remain comparable to those of our initial report, this confirming that haplo-HSCT from a family donor after selective depletion of αβ T cells and B cells represents an effective strategy for children with NMDs. Indeed, the number of diseases curable with an allogeneic HSCT increased rapidly in the recent years with the discovery of new disease-causing genes through next-generation sequencing.30 Thus, a “one-fits-all” solution, namely a transplant strategy virtually available for every NMD pediatric patient needing an allograft, is an attractive option.
Survival of our children affected by PID was remarkably high without significant differences between SCID and non-SCID patients. This is a major advancement since OS after haplo-HSCT performed using a positive selection of CD34+ cells resulted in survival as low as 47% in the latter population.31 Regarding SCID patients, the good outcomes reported are probably in part due to the short time period elapsing from diagnosis to HSCT, underlying the importance of a readily accessible transplant option. OS for SCID patients is superimposable to that recently published by The Primary Immune Deficiency Treatment Consortium on 100 patients with typical SCID or with leaky SCID, Omenn syndrome, or reticular dysgenesis given HSCT from different donors, including HLA-identical siblings, MUD, cord blood unit, and haploidentical relatives.32 In that study, the 2-year OS was 90%; notably, many patients were diagnosed with neonatal screening and, therefore, transplanted without active infections. As previously reported, active or recent infections impacted negatively on survival,31 this observation supporting the concept of rapidly proceeding to this kind of HSCT (or from other alternative donors) if an unaffected HLA-identical family donor is not available. As previously reported,9-13,33 after the selective removal of TCRαβ+ cells, the incidence of both acute and chronic GVHD remains remarkably low despite the fact that we did not administer any post-HSCT pharmacological prophylaxis. The low incidence of acute and chronic GVHD is particularly important in NMDs, in which GVHD occurrence does not determine any favorable effect (ie, graft-versus-leukemia effect), and, on the contrary, may contribute to the occurrence of more severe complications such as posttransplantation malignancies. In line with this observation, we recently reported a low incidence of GVHD, resulting in excellent outcomes in 24 FA patients treated with this type of haplo-HSCT platform.34 Moreover, the GRFS probability, which is used as a surrogate for quality of life, is remarkably high in our cohort.
GF (either primary or secondary) was shown to be the most challenging obstacle to be overcome in this study, particularly in a subgroup of patients at risk for this complication (namely, those affected by HLH, thalassemia, SAA, or osteopetrosis). The incidence of GF we observed is higher than that reported after T-replete haploHSCT with posttransplant cyclophosphamide (PT/Cy),35 as expected after the removal of αβ T cells that may counterbalance the lytic effect displayed by recipient lymphocytes against donor cells. We have identified several serum biomarkers that can be used for an early diagnosis,27,36 possibly allowing for preemptive interventions. In particular, since IFNγ seems to play a central role in GF (which resembles HLH features),37 emapalumab, a novel IFNγ-neutralizing monoclonal antibody proven effective in resistant/relapsing HLH,38 is being investigated as a preemptive treatment of GF in both malignant and nonmalignant settings [NCT04731298]. Since during the study period, busulfan, used for conditioning of patients with red blood cell disorders and HLH, was adjusted to maintain concentration at a steady state (Css) between 600 and 900 ng/mL, one may argue that increasing the target to 800-1050 ng/mL, as suggested by Bartelink and colleagues,39 might result into higher engraftment rate. Anyhow, a second transplant, mainly performed using the other parent as the donor, was able to rescue most of our patients experiencing GF, and the occurrence of GF did not influence the probability of OS.
Another major drawback of this approach is, as expected in the T-cell depleted (TCD) setting, the incidence of viral infections (while that of fungal infections is low, probably due to both the rapid neutrophil recovery and pharmacological prophylaxis we used against molds and yeasts), which carries significant morbidity and mortality. Thus, strategies aimed at reducing this risk are desirable. Regarding CMV, the most common viral threat after HSCT, the use of the new antiviral letermovir holds promises.40 However, several other viruses, such as ADV, are poorly controlled by pharmacological interventions. For this reason, adoptive infusion of virus-specific or nonspecific donor lymphocytes has been adopted.41 Regarding the latter strategy, a recent randomized trial testing the efficacy of repeated infusions of naïve-depleted (CD45RA-depleted) donor lymphocyte infusion (DLI) after TCRαβ+/CD19+-depleted haploHSCT failed to demonstrate a clinical effect in the study population.42 On the contrary, Roy and colleagues demonstrated a reduction of TRM in T-cell depleted haploHSCT setting with the use of allo-depleted DLI as compared with TCD-HSCT alone.43 We recently tested the use of genetically modified donor lymphocytes (transduced with the safety switch inducible Caspase-9, which can be activated in case of uncontrolled GVHD), showing feasibility and encouraging results in terms of survival,44 although specific analyses on viral infections are ongoing. Recovery of adaptive immunity after αβ T cell and B cell depleted haplo-HSCT is still suboptimal,29,45 once more calling for the implementation of new strategies aimed at accelerating immune reconstitution. Two of the 6 patients included in this study who did not reach independence from IgG replacement therapy had experienced acute GVHD, a well-known risk factor for this complication.46
The incidence of autoimmune complications we observed is in line with that reported by other authors.35 In 3 patients, the clinical course was characterized by multiple relapses of the autoimmune peripheral blood cytopenia, confirming the difficult-to-treat nature of this kind of post-HSCT complications.47 Notably, only 1 patient had mixed chimerism at the time of AIHA onset, this finding being different from what was reported by other studies, which suggested a correlation between mixed chimerism and development of autoimmune cytopenia.48
No formal comparison is available for other alternative treatment options, namely T-cell replete haploHSCT with PT/Cy and gene therapy. HaploHSCT with PT/Cy is a broadly used option since it is characterized by easiness of application. To date, several reports are available in the NMD setting,35,49-51 although many included less than 10 patients. Results from 2 larger studies, including 2735 and 73 patients,49 respectively (most of whom were affected by PIDs), show that this haplo-HSCT platform is characterized by a higher engraftment rate, but at the price of high incidence of both acute and chronic GVHD (ranging between 33%-46% and 16%-24%, respectively), which may affect quality of life in children with a long life expectancy. After initial problems related to malignant transformation of transduced HSC with retroviral vectors,52 gene therapy has evolved, reaching promising results in many disorders, including, among the others, ADA-SCID,53 X-SCID,54 WAS,55 and transfusion-dependent thalassemia.3 Its main advantage is related to the use of autologous cells, thus overcoming the problem of alloreactivity; moreover, despite the use of cytotoxic drugs to favor the engraftment of transduced cells, the grade of immunosuppression is far from that of an allogeneic HSCT, thus further reducing the infectious risks. However, results of this approach are still conditioned by disease-specific variables such as the hematopoietic reservoir for HSC transduction (eg, for IBMFS like Fanconi Anemia),56 the presence/absence of selective advantage of transduced cells, the number of transduced cells needed to revert the disease phenotype, and the safety profile, including the risk of developing clonal disorders.52 Also, HSCT carries a well-known risk of secondary malignancies.57 None of the patients reported suffering from this type of complication, although the follow-up is too short to draw firm conclusions on this risk.
TCRαβ+/CD19+ depleted haploHSCT compares favorably with other types of transplants from related58 and unrelated donors in the pediatric NMD setting.33,59 Moreover, despite being characterized by high initial costs, mainly related to the process and consumables to perform TCRαβ+/CD19+ depletion, a pharmacoeconomic analysis suggests that this approach may be cost-effective as compared with HSCT from MUD.60 Obviously, in cases of GF requiring a second HSCT, the pharmacoeconomic profile becomes less favorable.
In summary, our data obtained in a large cohort of pediatric patients confirm that this type of transplant is an attractive strategy for NMDs in view of the prompt availability of the donor, low incidence of TRM, as well as of both acute and chronic GVHD.
Acknowledgment
This work was partly supported by grants from the Associazione Italiana Ricerca sul Cancro (Investigator Grant ID 21724) (F.L.).
Authorship
Contributions: F.L. designed the study and supervised the project; P.M., F.G., E.B., and F.d.B. collected the data; P.M., M. Algeri, and F.L. analyzed and interpreted the data; P.M., D.P., F.G., D.A., R.M.P., A.B., A.M., L.S., E.B., M.D.N. F.d.B., M. Algeri, and F.L. were involved in the clinical management of patients; M. Andreani performed HLA typing; G.L. performed donor apheresis; G.L.P. performed graft manipulation and graft characterization; V.B., D.P., and M.F. performed immune monitoring; P.M., F.d.B., L.S., and F.L. wrote and edited the manuscript; and all authors had access to primary clinical trial data, contributed to the intellectual content of this article, and reviewed and approved the final manuscript.
Conflict-of-interest disclosure: P.M.: SOBI advisory board membership and consultancy outside the submitted work. F.L.: Amgen Speakers' Bureau and advisory board membership, Novartis Speakers' Bureau and advisory board membership, Bellicum Pharmaceuticals advisory board membership, Miltenyi Speakers' Bureau, Jazz Pharmaceutical Speakers' Bureau, Takeda Speakers' Bureau, Neovii advisory board membership, and Medac Speakers' Bureau outside the submitted work.
Correspondence: Pietro Merli, Department of Pediatric Hematology/Oncology, Cell and Gene Therapy, Bambino Gesù Children’s Hospital, Piazza S. Onofrio, 4, 00165 Rome, Italy; e-mail: pietro.merli@opbg.net.

