

Key Points
Patients with FL anti-HBc+ present with worse 10-year PFS and OS.
Patients with anti-HBc+ present with a higher frequency of ARID1A mutations and lack of EP300 mutations.
Abstract
Epidemiological studies have demonstrated the association between hepatitis B virus (HBV) infection and B-cell non–Hodgkin lymphoma (NHL), mainly for diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL). We studied a cohort of 121 patients with FL for HBV infection status, clinical features, and gene mutational profile. Anti-HBc was detectable in 16 patients (13.2%), although all had undetectable HBV DNA. Anti-HBcore+ (anti-HBc+) cases presented with older age at diagnosis than anti-HBc− cases (68.1 vs 57.2 years; P = .007) and higher β2-microglobulin (56.3% vs 28.9%; P = .04). All patients included in the study fulfilled criteria for treatment and received therapy with rituximab or rituximab-containing chemotherapy. There were no episodes of HBV reactivation or HBV hepatitis during treatment and/or maintenance. Remarkably, anti-HBc+ patients had significantly lower 10-year progression-free survival (PFS; 12.9% vs 58.3%; P < .0001) and overall survival (OS; 22.0% vs 86.2%; P < .0001), that remained at multivariate analysis. Gene mutational profiling of all cases showed that anti-HBc+ cases had higher incidence of ARID1A mutations and absence of EP300 mutations, 2 key epigenetic regulators in FL. Overall, our study shows that FL patients with resolved HBV infection have a worse outcome independently of other well-known clinical risk factors and a distinct gene mutational profile.
Introduction
Chronic hepatitis B infection is a global public health threat that causes considerable morbidity and mortality. Although hepatitis B virus (HBV) is an essentially hepatotropic DNA virus, it also exhibits the capacity to infect peripheral blood mononuclear cells, including non-Hodgkin lymphoma (NHL) cells.1,2 Besides being a risk factor for developing hepatocellular carcinoma, epidemiologic studies have suggested that chronic HBV infection can increase the risk of B-cell NHL, and it has been endorsed in a recent meta-analysis showing that HBV infection is significantly associated with diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL).3 Patients with occult HBV infection (presence of HBV DNA in liver and/or serum, with undetectable hepatitis B surface antigen [HBsAg]) and resolved HBV infection (HBsAg− and anti-HBcore+[anti-HBc+]) have been shown to contribute to the risk of developing NHL, although these situations are not considered in some studies because of the inadequacy of such information.3 In fact, exclusion of occult/resolved HBV infection might underestimate the association between HBV and B-cell NHL.4 Nevertheless, the mechanisms underlying HBV-induced NHL are still to be unraveled.
Only a few studies have evaluated the impact of HBV infection on the clinical course and prognosis of NHL.5 Patients with HBsAg+ DLBCL have been reported to have a more aggressive disease and shorter survival.6,7 Of note, these patients had increased mutation rate and a distinctive set of mutated genes.6 However, there is scarce information on patients with DLBCL with so-called resolved HBV infection. Moreover, the impact of chronic HBV infection in FL remains largely unknown. Here, we performed a clinical and genetic study on patients with FL in relation to HBV infection.
Methods
A cohort of 121 patients diagnosed with FL grade 1 to 3A was included in the study (supplemental data). Apart from standard staging studies, patients were prospectively screened for HBsAg, anti-HBc, hepatitis C virus (HCV), and HIV, and viral DNA was quantified if HBsAg or anti-HBc was detectable. All 121 patients included were white and from Spain or other European countries, and all fulfilled criteria for treatment according to the Groupe d'Etude des Lymphomes Folliculaires.8 Patients with HIV infection were excluded. Treatment consisted of rituximab (R), R plus CVP (R-CVP), R plus CHOP (R-CHOP), or R plus bendamustine (R-B), according to standard practice. In responding patients, maintenance with R was performed according to physician or patient preference. Before R treatment, patients with HBsAg+ or HBV DNA had to be started with nucleoside analogs (NAs), and patients with anti-HBc+ had the option of prophylactic NA or monitoring HBV DNA. Regardless of the strategy used, all these patients with anti-HBc+ were monitored for HBV DNA every 3 months up to 1 year beyond completion of R (as induction or maintenance).9 Tumoral FL mutational analysis was performed by next-generation sequencing through a DNA targeted custom panel of 64 FL-related genes implicated in epigenetics, BCR signaling, cell survival, immune response, mTORC1 pathway, and cell migration (supplemental methods and supplemental Table 1). The study was approved by the ethics committees of participating centers, and all patients provided written informed consent.
Results and discussion
In our FL cohort, the median age of the 121 patients was 59.3 years (interquartile range [IQR]: 48.6-69.1); 55.4% were men, and most of them had disseminated stage III to IV disease (91.7%). None of our patients presented HBsAg+; 16 (13.2%) were anti-HBc+ (all without detectable HBV DNA), and 5 (4.1%) had HCV positivity (3 in anti-HBc+ group). The number of patients with HCV was significantly higher in the anti-HBc+ (18.8% vs 2.0%; P = .025). Because patients with HBV infection have a risk of HBV reactivation when exposed to R, we have included HBsAg and anti-HBc in our workup of B-cell NHL since 2001.10 Prophylactic NA was given to 5 patients (entecavir in 2 and tenofovir in 3).
Baseline clinical characteristics according to anti-HBc status are shown in Table 1. Patients with anti-HBc+ displayed older age (mean age, 68.1 [IQR: 60.8-76.95] vs 57.2 [IQR: 47.7-68.09] years; P = .003). There were no significant differences in treatment schema used between groups, and no differences were observed in the complete response rate according to anti-HBc status. In our cohort, there were no episodes of HBV reactivation or HBV hepatitis. However, a post hoc analysis of the GOYA and GALLIUM trials has shown a risk of HBV reactivation of ∼10% if no prophylaxis is administered for DLBCL or FL with obinutuzumab or R-based treatment. Of note, >60% of cases included in this subanalysis was from Asian countries.11 Our data suggest that both prophylactic NA and strict monitoring are safe strategies in the real-world setting, at least in western countries.
Demographics and clinical characteristics of patients according to anti-HBc status
| Anti-HBc+ | Anti-HBc− | ||
|---|---|---|---|
| n = 16 (13.2%) | n = 105 (86.8%) | ||
| Follow-up duration, mean (IQR), mo | 107 (66-146) | 102 (59-140) | .839 |
| Age >60 y | 13 (81.3) | 46 (43.8) | .007 |
| Sex, male | 8 (50) | 59 (56.2) | .788 |
| Ann Arbor stage III to IV | 13 (81.3) | 98 (93.3) | .128 |
| Performance status ECOG 2 to 4 | 0 | 8 (7.6) | .595 |
| B symptoms | 4 (25.0) | 31 (29.5) | 1 |
| Bone marrow involvement | |||
| No | 5 (31.3) | 47 (44.8) | .278 |
| Yes | 11 (68.8) | 50 (47.6) | |
| Unknown | 0 | 8 (7.6) | |
| Increased LDH* | 3 (18.8) | 18 (17.3) | 1 |
| Hemoglobin <12 g/dL | 4 (25) | 22 (21) | .746 |
| Increased B2-microglobulin† | 9 (56.3) | 28 (28.9) | .044 |
| FLIPI score‡ | |||
| 0 to 1 | 2 (12.5) | 21 (20.8) | .392 |
| 2 | 5 (31.3) | 43 (42.6) | |
| 3 to 5 | 9 (56.3) | 37 (36.6) | |
| M7-FLIPI score† | |||
| Low | 11 (68.8) | 80 (79.2) | .346 |
| High | 5 (31.3) | 21 (20.8) | |
| Histological grade | |||
| 1 to 2 | 9 (56.3) | 55 (52.4) | .759 |
| 3a | 4 (25.0) | 35 (33.3) | |
| Undetermined | 3 (18.8) | 15 (14.3) | |
| Ki67 >30%§ | 5 (31.3) | 24 (27.9) | 1 |
| Induction regimen | |||
| R | 1 (6.3) | 10 (9.5) | .125 |
| R plus CVP/CHOP | 14 (87.5) | 66 (62.9) | |
| R plus bendamustine | 1 (6.3) | 29 (27.6) | |
| Response to primary treatment (complete response)† | 13 (81.3) | 86 (82.7) | 1 |
| Maintenance R | 12 (75) | 78 (74.3) | 1 |
| Anti-HBc+ | Anti-HBc− | ||
|---|---|---|---|
| n = 16 (13.2%) | n = 105 (86.8%) | ||
| Follow-up duration, mean (IQR), mo | 107 (66-146) | 102 (59-140) | .839 |
| Age >60 y | 13 (81.3) | 46 (43.8) | .007 |
| Sex, male | 8 (50) | 59 (56.2) | .788 |
| Ann Arbor stage III to IV | 13 (81.3) | 98 (93.3) | .128 |
| Performance status ECOG 2 to 4 | 0 | 8 (7.6) | .595 |
| B symptoms | 4 (25.0) | 31 (29.5) | 1 |
| Bone marrow involvement | |||
| No | 5 (31.3) | 47 (44.8) | .278 |
| Yes | 11 (68.8) | 50 (47.6) | |
| Unknown | 0 | 8 (7.6) | |
| Increased LDH* | 3 (18.8) | 18 (17.3) | 1 |
| Hemoglobin <12 g/dL | 4 (25) | 22 (21) | .746 |
| Increased B2-microglobulin† | 9 (56.3) | 28 (28.9) | .044 |
| FLIPI score‡ | |||
| 0 to 1 | 2 (12.5) | 21 (20.8) | .392 |
| 2 | 5 (31.3) | 43 (42.6) | |
| 3 to 5 | 9 (56.3) | 37 (36.6) | |
| M7-FLIPI score† | |||
| Low | 11 (68.8) | 80 (79.2) | .346 |
| High | 5 (31.3) | 21 (20.8) | |
| Histological grade | |||
| 1 to 2 | 9 (56.3) | 55 (52.4) | .759 |
| 3a | 4 (25.0) | 35 (33.3) | |
| Undetermined | 3 (18.8) | 15 (14.3) | |
| Ki67 >30%§ | 5 (31.3) | 24 (27.9) | 1 |
| Induction regimen | |||
| R | 1 (6.3) | 10 (9.5) | .125 |
| R plus CVP/CHOP | 14 (87.5) | 66 (62.9) | |
| R plus bendamustine | 1 (6.3) | 29 (27.6) | |
| Response to primary treatment (complete response)† | 13 (81.3) | 86 (82.7) | 1 |
| Maintenance R | 12 (75) | 78 (74.3) | 1 |
Values are reported as n (%) of patients unless indicated otherwise. Percentages might not add up to 100% because of rounding.
CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; CVP, cyclophosphamide, vincristine, and prednisone; ECOG, Eastern Cooperative Oncology Group; FLIPI, Follicular Lymphoma International Prognostic Index; LDH, lactate dehydrogenase
One case not known.
Eight cases not known.
Four cases not known.
Nineteen cases not known/not evaluated.
After a median follow-up of 94.3 months, 10-year progression-free survival (PFS) was 49.3% (95% confidence interval [CI], 40.0-60.4; 13 events overall), and 10-year overall survival (OS) was 75.0% (95% CI, 63.6-83.2; 11 deaths overall). Patients with anti-HBc+ showed significantly worse PFS than those in the anti-HBc− group, with 10-year PFS of 12.9% (95% CI, 1.6% to 36.1%) vs 58.3% (95% CI, 45.6% to 69.1%; P < .0001) (Figure 1A). Removal of the 3 double-positive cases (anti-HBc+/HCV+) did not affect these findings. Eleven patients relapsed in the anti-HBc+ group, and only one of the 5 biopsied cases showed transformation. The proportion of cases not responding or progressing in the 24 months after initiating treatment ( POD24) was significantly higher in patients with anti-HBc+ (43.8% vs 14.4%; P = .011). Accordingly, patients with anti-HBc+ had a significantly worse 10-year OS (22.0% [95% CI, 4.9% to 46.8%] vs 86.2% [95% CI, 75.8% to 92.5%]; P < .0001) (Figure 1B). Causes of death in the 11 patients among the anti-HBc+ group were 6 (55%) lymphoma progression; 2 (18%) second neoplasms; 2 (18%) HCV-related; and 1 (9%) cognitive impairment. We performed a multivariate analysis with other relevant factors for survival in FL, and anti-HBc+ remained statistically significant (supplemental Tables 2 and 3).
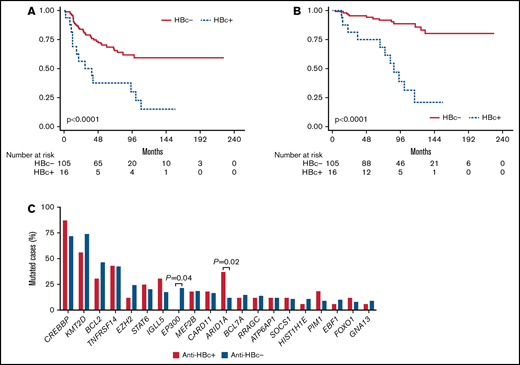
Outcome and mutation frequency according to anti-HBc. PFS (A) and OS (B) according to anti-HBc status. (C) Top 20 mutated genes and frequencies according to anti-HBc status; significant differences are marked with the P value.
Outcome and mutation frequency according to anti-HBc. PFS (A) and OS (B) according to anti-HBc status. (C) Top 20 mutated genes and frequencies according to anti-HBc status; significant differences are marked with the P value.
Gene mutational analysis of FL samples by targeted next-generation sequencing revealed an average of 7.3 mutations (range, 4-13) in anti-HBc+ cases and 5.5 mutations in anti-HBc− (range, 2-23; differences nonsignificant). The process of HBV DNA integration into the host genome is still unclear, but it probably enhances genomic instability, because HBV-related tumors, including hepatocellular carcinoma and HBsAg+ DLBCL, generally present with a higher rate of chromosomal abnormalities and mutations as shown by whole-exome sequencing.6,12,13 In our study, molecular profiling was based on a panel of 64 FL-associated genes, which limits the detection of such differences.
For the whole cohort, the most commonly mutated genes and their frequencies were in agreement with previous data in FL.14,15 Interestingly, patients with anti-HBc+ presented significantly different mutation frequencies in 2 genes involved in epigenetics when compared with anti-HBc− cases: a higher proportion (threefold) of mutations in ARID1A (37.5 vs 12.4%, respectively; P = .02) and lack of EP300 mutations (0% vs 29.1%; P = .04) (Figure 1C; supplemental Table 3). Knowledge of the complex network of interactions that HBV engages with its host is still limited, but accumulating evidence indicates that epigenetic modifications occurring in the course of infection in both the viral DNA and the host genome are essential to modulate viral activity and likely contribute to pathogenesis and cancer development.16 ARID1A encodes a subunit of the Switch/Sucrose-Nonfermentable (SWI/SNF) chromatin remodeling complex, which modulates numerous processes that require access to DNA, such as transcription, DNA damage repair, and replication. ARID1A mutations, mainly truncating, affect 10% to 15% of patients with FL and are included in the m7-FLIPI clinicogenetic risk model, although their prognostic impact is still unclear. Notably, HBV-related hepatocellular carcinomas are also enriched in ARID1A mutations, but the precise role of these alterations beyond chromatin accessibility remains to be elucidated.17 On the other hand, EP300 encodes a histone-acetyltransferase mutated in ∼15% of germinal center B-cell lymphomas. Together with its homologous CREBBP, both histone-acetyltransferases are known to be recruited and to interact with the hepatitis B viral DNA, in order to evade host defenses and to promote virus survival and persistence.18 Apart from the lack of mutations in EP300, patients with anti-HBc+ presented higher frequency of mutations in CREBBP (87.5 vs 71.4%, ns.), which might be preferentially affected in these cases, because alterations in both genes are generally mutually exclusive. Previous data regarding host mutations in HBV-related NHL are very scarce. Ren and colleagues explored the genetic landscape of HBV-associated DLBCL and observed, among other differences, an increase in ARID1A mutations and absence of EP300 mutations, in agreement with our findings.6
In summary, our study shows that patients with FL with resolved HBV infection have a worse outcome independently of other well-known clinical risk factors. In addition, anti-HBc+ cases show a distinct molecular profile with higher incidence of ARID1A alterations and lack of EP300 mutations. Further studies are needed to clarify our understanding on the pathogenesis of HBV in patients with FL.
Acknowledgments
This study was supported in part by Instituto de Salud Carlos III FIS-FEDER PI15/0459, FIS-FEDER PI19/00034, GILEAD GLD18/00117, 2017SGR205, PT20/00023, and Xarxa de Banc de Tumors de Catalunya sponsored by Pla Director d'Oncologia de Catalunya. The authors also acknowledge the Biobank of the Fundación MD Anderson International supported by the Instituto de Salud Carlos III grant PT17/0015/0008.
Authorship
Contribution: C.F.-R. designed and performed the research and the statistical analysis, analyzed and interpreted the results, and wrote the manuscript; J.J.R.-S. collected and analyzed the data and wrote the manuscript; J.G. performed the bioinformatic analysis; B.S.-G., L.B., J.M.S., J.F.G., R.D.-F., M.G.-R., E.G., and A.G. collected and analyzed the data; L.F.-I. and L.C. performed the research; L.C. performed histologic diagnostic and interpreted the results; B.B. designed the research, analyzed and interpreted the results, and wrote the manuscript; A.S. designed the research, performed the statistical analysis, analyzed and interpreted the results, and wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
Conflict-of-interest disclosure: A.S. has received research funding and speakers bureau fees from Roche; consultancy and speakers bureau fees from Janssen Pharmaceuticals; research funding from Gilead; and consultancy fees from Celgene. B.B. has received consultancy and speaker fees from ThermoFisher Scientific; consultancy and speaker fees from Qiagen; consultancy fees, speaker fees, and research funding from Roche; speaker and consultancy fees and research funding from Astra-Zeneca; speaker fees from Biocartis; speaker and consultancy fees from Merck-Serono; speaker and consultancy fees and research funding from Novartis; and speaker and consultancy fees from Bristol Myers Squibb. M.G.-R. has received research funding and lectures honoraria from Abbvie; research funding and lectures honoraria from Gilead Sciences; and consultancy fees from Intercept. B.S.-G. has received consultancy and speaker fees from Amgen; speaker fees from Gilead; consultancy and speakers fees from Novartis; speaker fees from Shire; and consultancy and speaker fees from Takeda. J.M.S. has received consultancy and speaker fees from Janssen; consultancy and speaker fees from Roche; consultancy and speaker fees from Gilead; consultancy and speaker fees from Celgene; consultancy fees from Bristol Myers Squibb; speaker fees from Takeda; consultancy and speaker fees from Novartis; and consultancy fees from Celltrion. E.G. has received consultancy and speaker fees from Janssen; consultancy and speaker fees from Abbvie; consultancy and speaker fees from Gilead; and consultancy and speaker fees from Roche. The remaining authors declare no competing financial interests.
Correspondence: Antonio Salar, Passeig Marítim 25-29, 08003 Barcelona, Spain; e-mail: ASalar@parcdesalutmar.cat.

