

TO THE EDITOR:
Primary central nervous system (CNS) lymphoma (PCNSL) is an aggressive non-Hodgkin lymphoma with CNS confinement; large B-cell lymphoma (LBCL) accounts for most PCNSL. The median age for PCNS-LBCL is 66 years, with occurrence in younger adults and pediatric cases being uncommon.1 Although immunodeficient children have an increased risk of developing PCNSL, most cases of pediatric PCNSL in the past few decades have been reported in immunocompetent individuals. Previous studies have reported that the prognosis for PCNS-LBCL is better in pediatric and younger adults than in older adults who have a median survival of <3 years.2,3 Aberrant constitutive activation of the NF-κB signaling pathway is a hallmark of PCNS-LBCL in adults and is characterized by frequent MYD88 mutation, CD79B mutation, PRDM1 mutation/deletion, CDKN2A biallelic deletion, and HLA gene cluster deletion.4-9 However, the molecular pathogenesis of PCNS-LBCL in children and young adults is largely unknown and may be different from that in older adults, helping to explain the divergent clinical outcomes.
This multi-institutional study was conducted to investigate the molecular characteristics of pediatric and young-adult PCNS-LBCL and define associations with clinical outcomes and pathologic features. The cohort consisted of 12 patients <40 years of age without known immunodeficiency, including 6 males and 6 females with median age at diagnosis of 17 years (range, 7-38 years). All patients were confirmed to have primary CNS confinement without systemic involvement based on staging positron-emission tomography/computed tomography and bone marrow biopsy. All patients were treated with a combination of dexamethasone, methotrexate, and rituximab, together with various accompanying agents (supplemental Tables 1-2). Tumors were uniformly composed of dense aggregates of large lymphoid cells with perivascular and parenchymal involvement, were Epstein-Barr virus (EBV) negative, and had diffuse strong immunoreactivity for B-cell markers (CD20 and PAX5). No BCL2 rearrangement and no MYC amplification or rearrangement were present (supplemental Tables 3-4). No appreciable differences in cytology or growth pattern were identifiable between these pediatric and young-adult patients with PCNS-LBCL compared with their older adult counterparts. The Institutional Review Board, Human Research Protection Program Committee at the University of California San Francisco approved this study (CHR 18-25787), which was conducted in accordance with the Declaration of Helsinki.
Targeted next-generation DNA sequencing and genome-wide copy number analysis were performed on the 12 tumors, as well as 6 conventional PCNS-LBCL from older adult patients (supplemental Methods). Based on the molecular signatures, 2 distinct groups were identified: “pediatric type, MYD88-wildtype” and “adult type, MYD88-mutant” (Figure 1). The pediatric type, MYD88-wildtype group consisted of 8 patients (5 males, 3 females) with a median age of 14 years (range, 7-25 years). No patients were immunocompromised or HIV positive, and none had a known tumor predisposition syndrome. Two tumors were supratentorial, 1 was infratentorial, and 2 were both supratentorial and infratentorial. Four cases were of the germinal center, and 4 were of the nongerminal center phenotype, according to Hans criteria.10 All tumors had a non–double-protein expressor phenotype, and none showed a double- or triple-hit genotype for MYC/BCL2/BCL6. The median Ki-67 labeling index was 90% (range, 40% to 95%). Genetic alterations commonly occurring in adults with PCNS-LBCL (MYD88 mutation, CDKN2A/B homozygous deletion, HLA gene cluster deletion, and PRDM1 mutation/deletion) were rare or absent (supplemental Figure 1; supplemental Tables 5-7). Instead, mutations in GNA13 (50%), NFKBIE (50%), and TP53 (75%) were recurrently identified. Although this group was highly enriched for pediatric age (88%), 25-year-old patient 4 was included based on the lack of MYD88 mutation and presence of NFKBIE and TP53 mutations. A single tumor in patient 15 showed GATA2 homozygous deletion, which has not been reported in systemic or PCNS-LBCL. Interestingly, alterations in GATA2 are recurrent in pediatric myeloid malignancies such as leukemia and myelodysplastic syndrome.11-14 Other likely oncogenic alterations in individual tumors included focal high-level amplification of JAK2 and CD274 (PD-L1) and EZH2 p.Y646N hotspot mutation that is a recurrent variant found in systemic LBCL and follicular lymphoma. Four tumors in this group exhibited a small number of chromosomal gains or losses (<5 per tumor), whereas the quantity of chromosomal aberrations was high (≥5) in the remaining 4 tumors. No differences in radiologic or histologic features between these pediatric-type PCNS-LBCL and their adult type counterparts was appreciated (Figure 2A-F).

Genomic analysis identifies a unique pediatric subtype of EBV-negative primary CNS large B-cell lymphoma. Clinical, histologic, and genetic features of the 12 pediatric and young-adult patients with PCNS-LBCL, alongside 6 conventional cases of PCNS-LBCL occurring in older adults. Categorization into pediatric- and adult-type tumor groups was performed according to recurrent molecular alterations involving MYD88, PRDM1, CDKN2A/B, HLA gene cluster, NFKBIE, GNA13, and TP53.
Genomic analysis identifies a unique pediatric subtype of EBV-negative primary CNS large B-cell lymphoma. Clinical, histologic, and genetic features of the 12 pediatric and young-adult patients with PCNS-LBCL, alongside 6 conventional cases of PCNS-LBCL occurring in older adults. Categorization into pediatric- and adult-type tumor groups was performed according to recurrent molecular alterations involving MYD88, PRDM1, CDKN2A/B, HLA gene cluster, NFKBIE, GNA13, and TP53.
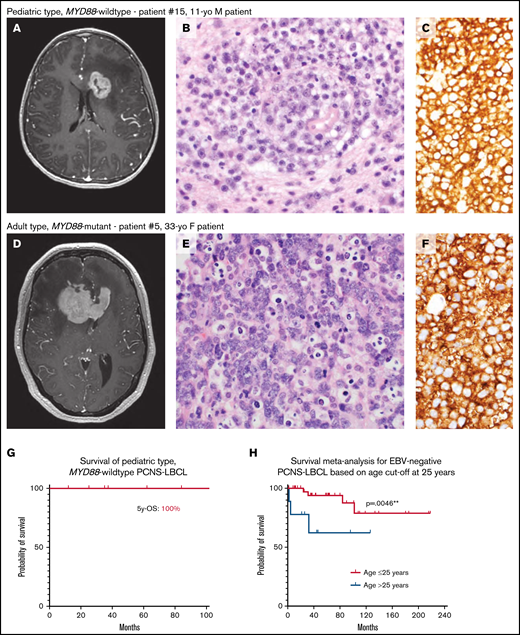
Pediatric MYD88-wildtype PCNS-LBCL is associated with favorable clinical outcomes compared with its adult-type counterpart. (A-F) Representative radiologic and histopathologic findings in pediatric- and adult-type PCNS-LBCL. Preoperative T1-weighted postcontrast magnetic resonance images of patients with pediatric type, MYD88-wildtype (A) or adult type, MYD88-mutant (D) PCNS-LBCL, showing overlapping radiologic features, including intraparenchymal mass lesions with diffuse enhancement and significant edema in the adjacent white matter. Histologic analysis showed sheets of malignant large B-cells diffusely expressing CD20. Hematoxylin and eosin, magnification ×400 (B,E); and CD20, original magnification, ×400 (C,F). (G) Kaplan-Meier survival plot of 7 patients with pediatric type, MYD88-wildtype PCNS-LBCL. (H) Kaplan-Meier meta-analysis of 53 pediatric and young-adult patients with PCNS-LBCL stratified by age ≤ 25 years vs 26 to 40 years.
Pediatric MYD88-wildtype PCNS-LBCL is associated with favorable clinical outcomes compared with its adult-type counterpart. (A-F) Representative radiologic and histopathologic findings in pediatric- and adult-type PCNS-LBCL. Preoperative T1-weighted postcontrast magnetic resonance images of patients with pediatric type, MYD88-wildtype (A) or adult type, MYD88-mutant (D) PCNS-LBCL, showing overlapping radiologic features, including intraparenchymal mass lesions with diffuse enhancement and significant edema in the adjacent white matter. Histologic analysis showed sheets of malignant large B-cells diffusely expressing CD20. Hematoxylin and eosin, magnification ×400 (B,E); and CD20, original magnification, ×400 (C,F). (G) Kaplan-Meier survival plot of 7 patients with pediatric type, MYD88-wildtype PCNS-LBCL. (H) Kaplan-Meier meta-analysis of 53 pediatric and young-adult patients with PCNS-LBCL stratified by age ≤ 25 years vs 26 to 40 years.
The adult type, MYD88-mutant group consisted of 4 young-adult patients (25-38 years of age) and the 6 cases of conventional PCNS-LBCL that occurred in older adults. All tumors harbored activating hotspot missense mutations in MYD88. In addition, 80% demonstrated homozygous deletion of CDKN2A/B, 60% demonstrated inactivating mutations or deletion of PRDM1, 60% demonstrated focal homozygous deletions of the HLA gene cluster on chromosome 6p22.1, and 40% demonstrated CD79B mutations. Only single tumors contained TP53, GNA13, or NFKBIE mutations (10% frequency each), in contrast to their frequent occurrence in the pediatric type tumors. Most adult-type, MYD88-mutant tumors (80%) had a high number of chromosomal copy number gains or losses (≥5).
A Kaplan-Meier survival analysis demonstrated a 5-year overall survival (OS) of 100% for the pediatric-type, MYD88 wild-type group, which was substantially longer than PCNS-LBCL in adult patients in previous studies15-20 (Figure 2G; supplemental Tables 8-9). We next performed meta-analysis of clinical outcome data, including 37 additional patients with PCNS-LBCL aged <40 years from 5 published studies,2,21-24 along with 5 additional previously unpublished cases. In a total cohort of 53 immunocompetent children and young adults with clinically and pathologically confirmed EBV− PCNS-LBCL, patients aged ≤25 years at the time of initial diagnosis had significantly better OS compared with those >25 years of age (P = .0046; Figure 2H). The respective 5- and 10-year OS estimates for patients aged ≤25 years were 93.8% and 78.8%, which were substantially longer than those for young adults aged 26 to 40 years (62.2% at both intervals) and also older adults from multiple prior large outcome studies.
To our knowledge, this is the first study to interrogate the genomic landscape of EBV-negative PCNS-LBCL in children and adolescents. We discovered a unique pediatric type, MYD88-wildtype PCNS-LBCL that correlated with age of onset ≤25 years and was genetically defined by enrichment for TP53, NFKBIE, and GNA13 mutations, along with an absence of MYD88 mutation, CDKN2A/B homozygous deletion, and HLA gene cluster deletion and a paucity of CD79B and PRDM1 mutations. Young adults between 25 and 40 years of age had genetic profiles similar to those of conventional PCNS-LBCL in older adults. We speculate that these may represent a tumor type that is biologically equivalent to that most commonly occurring in older adults, and we thus collectively termed this group adult type, MYD88-mutant. Our findings suggest that unlike adult-type tumors, constitutive activation of the NF-ĸB pathway in pediatric-type PCNS-LBCL is driven by NFKBIE and GNA13 mutations instead of MYD88 hotspot mutations. The effects of the frequent TP53 mutations are likely to synergize with the prosurvival signals driven by NFKBIE and GNA13 mutations in pediatric-type PCNS-LBCL. The intact HLA alleles in the pediatric-type tumors may be associated with a unique tumor microenvironment and immune status correlating with better overall survival, in contrast with the MYD88-mutant tumors in adults that have frequent deletions of the HLA gene cluster associated with immune evasion.5,9
In summary, we have identified a genetically distinct subtype of EBV-negative PCNS-LBCL occurring in immunocompetent pediatric and young-adult patients ≤25 years of age. This MYD88-wildtype group harboring frequent TP53, NFKBIE, and GNA13 mutations has favorable outcomes compared with the MYD88-mutant group occurring in older adults >25 years of age. Prospective next-generation DNA sequencing is advocated for PCNS-LBCL in the pediatric and young-adult population, for clinically distinguishing this unique pediatric-type disease.
Acknowledgments: The authors thank the staff of the UCSF Clinical Cancer Genomics Laboratory for assistance with genetic profiling.
This study is supported by the Clinical Research Endowment Grant and Rebecca Frankel Award from the University of California San Francisco, Department of Pathology to K.W.W. D.A.S. is supported by the Morgan Adams Foundation and the NIH Director’s Early Independence Award (DP5 OD021403). J.L.R. is supported by Leukemia and Lymphoma Society and NIH R01CA139-83-01A1 and R01CA239462 grants. C.-H.G.L. is supported by the UCSF Training Program in Translational Brain Tumor Research, National Institutes of Health (NIH), National Cancer Institute Grant T32CA151022.
Contribution: E.G., C.-H.G.L., D.A.S., and K.W.W. designed the study, performed experiments, and contributed to specimen collection, clinical and pathologic data collection, pathologic review, data analysis, and statistical analysis and wrote the manuscript; D.A.S. and K.W.W. supervised the study; R.S.O. contributed to review of the pathology; R.Z., D.R.B., K.S., G.B.W., S.D., A.B., A.P., and T.T. contributed to specimen collection, data collection, and review of the pathology; Z.Q. and J.Y. performed and evaluated the FISH; J.L.R., L.G.-R., A.A., M.J.B., O.A., A.L.C., J.S.M., and S.K. provided clinical data; M.P. contributed to data collection, pathologic review, and statistical analysis; and all authors had access to clinical, pathologic, and molecular data, critically reviewed the manuscript, and approved its submission.
Conflict-of-interest disclosure: S.K. receives consulting fees from Incyte Corporation and Karyopharm Pharmaceutics. The remaining authors declare no competing financial interests.
Correspondence: Kwun Wah Wen, Department of Pathology, University of California, San Francisco, 505 Parnassus Ave, M545, Box 0102, San Francisco, CA 94117; e-mail: kwun.wen@ucsf.edu.

