

Key Points
One intravenous dose of oncolytic VSV has single-agent activity in patients with relapsed refractory T-cell lymphoma.
Direct oncolytic tumor cell killing and subsequent immune cell activation were observed, resulting in durable remissions in responding patients.

Abstract
Clinical success with intravenous (IV) oncolytic virotherapy (OV) has to-date been anecdotal. We conducted a phase 1 clinical trial of systemic OV and investigated the mechanisms of action in responding patients. A single IV dose of vesicular stomatitis virus (VSV) interferon-β (IFN-β) with sodium iodide symporter (NIS) was administered to patients with relapsed/refractory hematologic malignancies to determine safety and efficacy across 4 dose levels (DLs). Correlative studies were undertaken to evaluate viremia, virus shedding, virus replication, and immune responses. Fifteen patients received VSV-IFNβ-NIS. Three patients were treated at DL1 through DL3 (0.05, 0.17, and 0.5 × 1011 TCID50), and 6 were treated at DL4 (1.7 × 1011 TCID50) with no dose-limiting toxicities. Three of 7 patients with T-cell lymphoma (TCL) had responses: a 3-month partial response (PR) at DL2, a 6-month PR, and a complete response (CR) ongoing at 20 months at DL4. Viremia peaked at the end of infusion, g was detected. Plasma IFN-β, a biomarker of VSV-IFNβ-NIS replication, peaked between 4 hours and 48 hours after infusion. The patient with CR had robust viral replication with increased plasma cell-free DNA, high peak IFN-β of 18 213 pg/mL, a strong anti-VSV neutralizing antibody response, and increased numbers of tumor reactive T-cells. VSV-IFNβ-NIS as a single agent was effective in patients with TCL, resulting in durable disease remissions in heavily pretreated patients. Correlative analyses suggest that responses may be due to a combination of direct oncolytic tumor destruction and immune-mediated tumor control. This trial is registered at www.clinicaltrials.gov as #NCT03017820.
Introduction
Oncolytic virotherapy (OV) is an experimental immunomodulatory approach to the treatment of cancer that exploits the ability of certain viruses to propagate selectively at sites of tumor growth.1,2 Local intratumoral spread of the oncolytic virus infection leads to inflammatory killing (oncolytic phase) of virus-infected tumor cells, provoking and amplifying adaptive T-cell responses (immune phase) against viral- and tumor-associated antigens, which, if sufficiently powerful, can mediate long-term tumor control.3,4 This dual mechanism of action comprising oncolytic and immune phases of tumor cell killing was demonstrated in patients with advanced malignant melanoma who experienced local and distant tumor responses after intratumoral administration of Talimogene laherparepvec, which gained marketing approval in 2015.5,6
Moving beyond intratumoral OV delivery, systemic therapy is now being more aggressively pursued to deliver OV to multiple metastatic tumor deposits and diminish reliance on the limited abscopal immune effects of intratumoral therapy. However, despite encouraging outcomes in mouse models, human trials of single agent systemic OV have so far met with only anecdotal success.7,8 Potential factors contributing to the failure of intravenously administered OVs to impact human cancer include (1) immediate virus dilution in ∼5 L of blood, (2) rapid virus inactivation by preformed antiviral antibodies and/or sequestration by the reticuloendothelial system, and (3) poor virus extravasation across the walls of tumor blood vessels.9-12
Vesicular stomatitis virus (VSV), a bullet-shaped rhabdovirus, is highly attractive as an oncolytic virus with well-documented antitumor activity in animal models.13-15 First, VSV can be manufactured at very high titers suitable for intravenous dosing. Second, VSV naturally causes a self-limited blistering illness in cloven animals but does not naturally infect humans; hence, it has very low seroprevalence in patients with cancer. Third, with a diameter of only 70 nm, the bullet-shaped VSV particles can extravasate efficiently from leaky tumor neovessels compared with several other popular OV platforms.16 We therefore generated VSV interferon-β (IFN-β) NIS, an oncolytic VSV that was engineered to encode both IFN-β and the thyroidal NIS to boost its tumor specificity, proinflammatory activity, and in vivo trackability.17 IFN-β confers specificity by limiting VSV spread in normal cells, has antitumor activities, and enhances dendritic cell-driven antitumor T-cell responses.18 Expression of NIS in the infected cell allows uptake of radiotracers for single photon emission computed tomography (SPECT) or positron emission tomography (PET) imaging of sites of virus infection.19 VSV-IFNβ-NIS was manufactured and subjected to comprehensive pharmacological and toxicological testing in rodents, canines with spontaneous cancers, and swine, a natural host species before phase 1 clinical testing.20-23
Herein, we describe the results of the first in-human phase 1 dose escalation clinical trial of IV administration of a single dose of VSV-IFNβ-NIS in 15 patients with relapsed refractory hematological malignancies. Overall, there were no dose-limiting toxicities with compelling evidence of dose-dependent clinical activity, particularly in patients with T-cell lymphoma (TCL).
Methods
Patient eligibility criteria
The protocol was approved by the Mayo Clinic Institutional Review Board. Adult patients with relapsed or refractory multiple myeloma (MM), TCL, or acute myeloid leukemia (AML) were included. Patients should have <15% plasma cells for myeloma or plasmacytoma <5 cm in largest diameter, TCL with mass <5 cm, and AML with <30% circulating blasts and <50% bone marrow blasts. Patients with acute promyelocytic leukemia (rearranged AML M3) were excluded.
Study design
This was a single center phase 1 trial to determine the maximum tolerated dose of VSV-IFNβ-NIS in patients with relapsed refractory MM, TCL, and AML. The secondary objectives were to determine the safety profile and estimate clinical response rate of VSV-IFNβ-NIS. Correlative objectives included monitoring viral replication through plasma IFN-β transgene levels and SPECT/CT imaging using the NIS gene, measuring viral genomes through quantitative reverse transcription (qRT) polymerase chain reaction (PCR) of VSV RNA and determining virus shedding (if any) in body fluids. The doses tested were 5 × 109 TCID50 (50% tissue culture infectious dose), 1.7 × 1010, 5 × 1010, and 1.7 × 1011 TCID50. The virus was administered by IV infusion over 30 minutes. All patients received 650 mg acetaminophen and 50 mg diphenhydramine hydrochloride before infusion and every 6 hours for 24 hours to mitigate fever and hypotension.
Safety and response assessments
All patients were available for toxicity evaluation. Adverse events (AEs) were determined by the Common Terminology Criteria for Adverse Events (CTCAE) version 4 criteria except for cytokine release syndrome (CRS) grading, which was graded according to the 2014 Lee criteria.24 Dose escalation and tolerability were based on the rules of a standard 3 + 3 phase 1 trial design.25 Response assessment was made as per International Myeloma Working Group response criteria for MM,26 by the Lugano Classification for patients with TCL,27 and according to European LeukemiaNet response criteria for patients with AML.28
NIS imaging
All patients had planar and SPECT/CT with 20 mCi 99mTc-pertechnetate, given 30 minutes before NIS imaging.
Pharmacodynamic and pharmacokinetic analyses
For assessment of viremia and viral shedding, RNA was isolated from whole blood collected in Paxgene RNA tubes, mouth rinse, buccal swabs, and urine samples. Quantitative RT-PCR was performed to determine VSV-nucleocapsid (N) RNA copy number. Samples were also cocultured on Vero cells for virus isolation to assess viral shedding.20 Anti-VSV antibodies was evaluated using standard plaque reduction neutralization assay. Plasma cytokine levels were measured using a 30-plex human cytokine kit (Invitrogen LHC6003M). A human IFN-β enzyme-linked immunosorbent assay (PBL Assay Science, #41415) kit was used to quantify plasma IFN-β levels.
Statistical analysis
Incidence of dose-limiting toxicities as well as AEs were tabulated by DL. AEs were summarized by type as well as maximum grade and duration across patients in each DL. Spearman rank correlation tests were used to evaluate correlation between laboratory-based measures, and Wilcoxon rank sum tests were used to compare these continuous measures between response groups. Response was also summarized by DL and disease group, in which estimated response rates were calculated along with corresponding 95% binomial confidence intervals.
Results
Patients and treatments
The protocol and informed consent forms were reviewed and approved by the Mayo Clinic Institutional Review Board and Biosafety committee. Between April 2017 and August 2020, 15 patients with relapsed refractory MM (n = 7), TCL (n = 7), or AML (n = 1) consented to the study and were treated with 1 IV infusion of VSV-IFNβ-NIS across 4 DLs (Figure 1A; supplemental Table 1). The median age was 64 years (range, 33-88 years), 60% were male, with Eastern Cooperative Oncology Group 0 and 1 in 66.7% and 33.3%, respectively. The median prior line of systemic cancer therapies was 5 (range, 2-11).
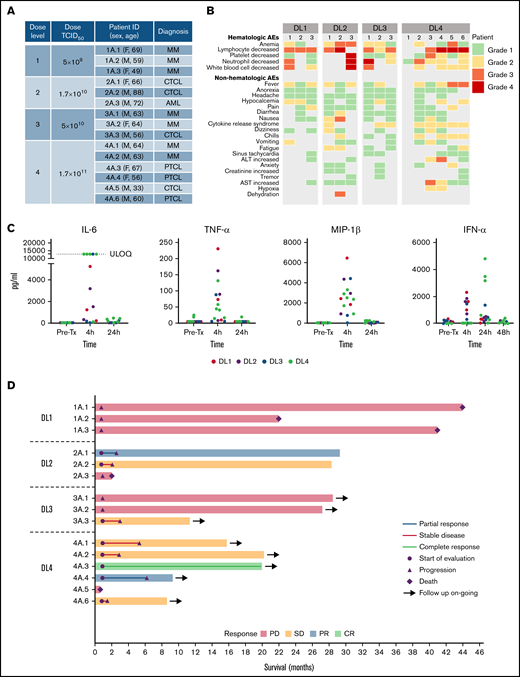
Clinical outcomes of treatment with 1 IV infusion of VSV-IFNβ-NIS. (A) Summary of patients treated in the study. (B) Maximum grade AEs by DL for each patient. (C) Levels of selected cytokines/chemokines before and after VSV infusion. (D) Swimmer plot (n = 15), in which each bar represents an individual patient as designated. Day 28 responses are indicated. Bars with solid arrows show patients with an ongoing response. ULOQ, upper limit of quantitation.
Clinical outcomes of treatment with 1 IV infusion of VSV-IFNβ-NIS. (A) Summary of patients treated in the study. (B) Maximum grade AEs by DL for each patient. (C) Levels of selected cytokines/chemokines before and after VSV infusion. (D) Swimmer plot (n = 15), in which each bar represents an individual patient as designated. Day 28 responses are indicated. Bars with solid arrows show patients with an ongoing response. ULOQ, upper limit of quantitation.
Safety
All 4 DLs were evaluated. No dose-limiting toxicities were observed, and 3 additional patients were treated at the top dose per protocol (n = 6 at DL4). The most common AEs observed were hematologic, typically returning to baseline within 2 to 10 days (Figure 1B). The 30-minute virus infusions were well tolerated in all patients at all DLs. A short-lived febrile response to the virus infusion was encountered in most patients, starting a few hours after infusion and resolving overnight, and was often associated with nausea, vomiting, hypotension, and/or mild hypoxemia. Multiplex cytokine analysis of plasma collected before therapy and after virus infusion revealed a transient elevation of multiple cytokines at the 4-hour timepoint, with return to baseline levels by 24 or 48 hours (Figure 1C). After reviewing the clinical and cytokine data with the US Food and Drug Administration, it was determined that this postinfusion reaction should be classified as CRS per Lee criteria.24,29 CRS (grades 1-2; Figure 1B) was most prominent at the highest DL and was observed in 5 of 6 patients at DL4. The CRS was transient and resolved in 24 to 48 hours. Patients were given supportive care with close monitoring; only 1 patient (patient 2A.1 at DL2) required transient support (4 hours) of 1 low-dose vasopressor (norepinephrine).
Clinical activity
The swimmer plot summarizing outcome of all patients is shown in Figure 1D. There were 3 objective responses, all in patients with TCL (Figure 1D). Interestingly, 5 of the 7 patients with TCL who were treated with VSV-IFNβ-NIS demonstrated mixed responses with regression of 1 or more tumors (supplemental Tables 1-2). The most compelling responses were seen at DL4, as detailed below (Figure 2).

A single dose of VSV-IFNβ-NIS monotherapy has clinical activity in patients with TCL. Photographs (A) and PET/CT scans (B) showing resolution of FDG-avid lesions of patient 4A.3 with TCL with cutaneous relapse of nodal PTCL who achieved a CR after receiving 1 dose of VSV-IFNβ-NIS at DL4. (C) FDG PET images of patient 4A.4 with nodal PTCL showing remarkable improvement in FDG-avid cervical, mediastinal, axillary, and abdominal lymph node disease (arrows) with decreased splenic size and FDG uptake (arrowheads) at 1 month. (D) Coronal fused FDG PET/CT images demonstrate improvement in splenic disease (arrowheads), with continued response in the spleen at the 6-month evaluation.
A single dose of VSV-IFNβ-NIS monotherapy has clinical activity in patients with TCL. Photographs (A) and PET/CT scans (B) showing resolution of FDG-avid lesions of patient 4A.3 with TCL with cutaneous relapse of nodal PTCL who achieved a CR after receiving 1 dose of VSV-IFNβ-NIS at DL4. (C) FDG PET images of patient 4A.4 with nodal PTCL showing remarkable improvement in FDG-avid cervical, mediastinal, axillary, and abdominal lymph node disease (arrows) with decreased splenic size and FDG uptake (arrowheads) at 1 month. (D) Coronal fused FDG PET/CT images demonstrate improvement in splenic disease (arrowheads), with continued response in the spleen at the 6-month evaluation.
Patient 4A.3, a 67-year-old woman with heavily pretreated multisite systemic anaplastic lymphoma kinase (ALK)-negative anaplastic large cell lymphoma that initially involved inguinal and iliac lymph nodes, returned to clinic with numerous rapidly relapsing fluorodeoxyglucose (FDG)-avid raised dermal and subdermal lesions on her distal left lower extremity (Figure 2A-B). She received a single infusion of VSV-IFNβ-NIS at DL4. She had received 5 prior lines of systemic anticancer therapy, including lenalidomide and CHOEP, gemcitabine and oxaliplatin, oral cyclophosphamide, nivolumab, and brentuximab vedotin, and was not a candidate for autologous stem cell transplantation. During the first 24 hours after virus infusion, her serum IFN-β level rose steeply to 18 213 pg/mL and declined slowly to baseline thereafter, providing a clear indication of intratumoral virus amplification. Three months after virus administration, all detectable lymphomatous lesions had completely regressed, and the complete response remains durable at 20 months in the absence of any additional antilymphoma therapies.
Patient 4A.4, a 56-year-old woman with diagnosis of multiply relapsed peripheral TCL (PTCL), returned to the clinic with rapidly progressing FDG-avid lymphadenopathy and FDG-avid splenomegaly (Figure 2C-D). Her prior therapies included CHOP, ifosfamide, carboplatin and etoposide (ICE) induction therapy, autologous stem cell transplantation with BEAM (Carmustine + Etoposide + Cytarabine + Melphalan) conditioning, ruxolitinib, romidepsin, pembrolizumab, and pralatrexate. She received a single IV infusion of VSV-IFNβ-NIS at DL4 and experienced a 6-month partial response. Twenty-four hours after virus infusion, her serum IFN-β level peaked at 662 pg/mL and declined slowly to baseline thereafter, suggesting robust intratumoral amplification of the virus. At her day 28 return visit, the PET/CT scan showed significant reduction in FDG activity of multiple previously seen nodes both above and below the diaphragm associated with >50% reduction in the sum of perpendicular diameters of her sentinel lesions (Figure 2C). There was also marked reduction in FDG-avid spleen uptake, with the splenic SUVmax and SUVmean decreased from 7.0 and 4.5 at baseline to 3.9 and 2.6, respectively, at day 28 follow-up (Figure 2D). The splenic total lesion glycolysis also decreased by >50%, from 1477.8 to 701.9 g and from 12.6 cm at baseline to 9.8 cm at day 28. Even at the 6-month scan, there was continued deepening response in the spleen, but by that time the patient had relapsing disease in the cervical lymph nodes.
Patient 2A.1, who was treated at DL2, had received 11 prior lines of systemic anticancer therapies for her mycosis fungoides (cutaneous TCL), presented with multifocal cutaneous lesions, and achieved a partial response lasting 3 months. Of the multiple skin lesions, 0.7 × 0.3 cm right eye, 2 × 1 cm right arm, 3 × 2 cm left arm, 8 × 5.5 cm right flank, and 4 × 2.5 cm medial right thigh lesions were measured at baseline (total surface area = 18.21 cm2). Twenty-three days after treatment with VSV-IFNβ-NIS, both the right eye and left arm lesions had completely resolved, the right arm lesion decreased to 1 × 1 cm, the right flank lesion retained the same dimensions but had a central area of clearing, and the right medial thigh lesion decreased to 3.5 × 1 cm (total surface area = 4.6 cm2). At the 3-month assessment, she relapsed with a single isolated right eye lesion but demonstrated continued response in the other lesions. The right eye lesion was subsequently irradiated (4 Gy) with deepening of responses of other nontarget lesions, lasting 2 years to next therapy, which suggested an augmented abscopal effect.
Although the numbers of patients enrolled in this phase 1 study were small, response heterogeneity was striking and some patients with TCL were entirely resistant to the oncolytic action of the virus. As an example, patient 4A.5 was a 33-year-old man with aggressive CD30+ mycosis fungoides who had extensive, bulky, treatment-refractory lymphomatous skin lesions that were highly glucose-avid on FDG PET/CT scan and were progressing rapidly (supplemental Figure 1). His prior therapies included brentuximab vendotin plus cyclophosphamide + doxorubicin + prednisone (CHP), gemcitabine, dexamethasone and cisplatin (GDP), ICE, romidepsin, mogamulizumab, total skin electron therapy, and pembrolizumab. After enrolling in the study, he received a single infusion of VSV-IFNβ-NIS at DL4. In contrast to other patients enrolled in the DL4 cohort, he had only a minor reaction to the virus infusion and did not develop CRS. Also, in contrast to other patients treated at DL4, his serum IFN-β level 24 hours after virus infusion was very low at 17 pg/mL (1000-fold lower than for patient 4A.3, who received the same virus dose), indicating that his cutaneous tumors were highly refractory to the virus infection. In keeping with these observations, there was no observable slowing of the growth of his cutaneous tumors after virus infusion, and he expired a few months later due to continued disease progression.
NIS imaging of VSV infection
The virus encodes the NIS transgene that, when expressed by the infected cell, concentrates 99mTc-pertechnetate and allows imaging of sites of virus infection.19 All patients underwent 99mTc-pertechnetate planar and SPECT/CT imaging at baseline and 24 hours after infusion, and 6 patients underwent additional 99mTc-pertechnetate imaging on day 5. A single patient with myeloma (patient 3A.1) had an osteolytic lesion in the right ilium that demonstrated increased 99mTc-pertechnetate uptake on days 1 and 5 after VSV treatment, confirming virus delivery and infection of the myeloma (Figure 3A). This same lesion demonstrated decreased FDG activity on comparison before and after therapy on PET/CT (Figure 3B), which was correlated with early tumor response, although the patient had overall disease progression. This contrasts with a left acetabular lesion in the same patient that demonstrated no increased uptake on the posttherapy SPECT/CT (Figure 3C), and at 6-month follow-up PET/CT, this lesion had increased in size, with progressive bone destruction (Figure 3D) and increased FDG uptake (Figure 3E). There were no other sites of increased 99mTc-pertechnetate uptake on postinfusion imaging in any other patient, including those with PR or CR.

Positive NIS imaging of virus-infected tumor corresponded with reduction in FDG avidity. (A) In patient 3A.1 with multiple myeloma, axial fused 99mTc-pertechnetate SPECT/CT images demonstrate increasing uptake within a lytic lesion in the anterior right ilium (arrows) on comparison at baseline before VSV, day 1 (24 hours), and day 5 images (arrows). (B) This same lesion demonstrated decreased FDG activity when comparing pre- and posttherapy PET/CT images (arrows). (C) In contrast, a left acetabular lesion in the same patient demonstrated no increased uptake on SPECT/CT at baseline, day 1, or day 5 scans, suggesting minimal VSV infection (arrows). The 6-month follow-up CT (D) and fused PET/CT images (E) showed progression of the acetabular lesion, with increased size, bone destruction, and FDG uptake (arrows).
Positive NIS imaging of virus-infected tumor corresponded with reduction in FDG avidity. (A) In patient 3A.1 with multiple myeloma, axial fused 99mTc-pertechnetate SPECT/CT images demonstrate increasing uptake within a lytic lesion in the anterior right ilium (arrows) on comparison at baseline before VSV, day 1 (24 hours), and day 5 images (arrows). (B) This same lesion demonstrated decreased FDG activity when comparing pre- and posttherapy PET/CT images (arrows). (C) In contrast, a left acetabular lesion in the same patient demonstrated no increased uptake on SPECT/CT at baseline, day 1, or day 5 scans, suggesting minimal VSV infection (arrows). The 6-month follow-up CT (D) and fused PET/CT images (E) showed progression of the acetabular lesion, with increased size, bone destruction, and FDG uptake (arrows).
Pharmacokinetics and pharmacodynamics
Quantitative RT-PCR was used to track presence of VSV-nucleocapsid (N) RNA in whole blood (viremia) and virus shedding (if any) in body fluids (mouth rinse, buccal swabs, urine). Viremia was short-lived, was highest at the end of infusion, and declined thereafter (Figure 4A). No secondary viremia was observed. There is a trend for higher viremia at 24 hours at the higher DLs (Figure 4B) but it is not statistically significant (P = .49; analysis of variance). Potential viral shedding into the buccal cavity or urine was also evaluated by qRT-PCR and virus recovery assays. VSV-N RNA was below levels of detection in most of the timepoints analyzed, except for very low levels at day 2 in the mouth rinse in some patients across DLs (supplemental Figure 2). Importantly, no infectious virus was isolated from any body fluids or buccal swabs at any time points from any patients, indicating no shedding of infectious virus (supplemental Figure 2).

Pharmacokinetics and pharmacodynamics of VSV-IFNβ-NIS after IV infusion. (A) Profile of viremia (VSV-N RNA in blood) in each patient measured at baseline, end of infusion, 30 minutes, 60 minutes, 4 hours, 24 hours, and 3, 8, 15, and 28 days after VSV. (B) 24-hour viremia levels of each patient at all DLs. Corresponding plasma IFN-β profile (C) and peak IFN-β levels (D) per DL. (E) Cell-free DNA in plasma of patients. (F) IFN-γ ELISPOT assay for T cells reactive against select tumor antigens, VSV-N, or cytomegalovirus, Epstein-Barr virus, and Flu virus positive control. (G) Anti-VSV neutralizing antibody titers (average of patients per DL). Mean ± standard error of the mean shown where applicable.
Pharmacokinetics and pharmacodynamics of VSV-IFNβ-NIS after IV infusion. (A) Profile of viremia (VSV-N RNA in blood) in each patient measured at baseline, end of infusion, 30 minutes, 60 minutes, 4 hours, 24 hours, and 3, 8, 15, and 28 days after VSV. (B) 24-hour viremia levels of each patient at all DLs. Corresponding plasma IFN-β profile (C) and peak IFN-β levels (D) per DL. (E) Cell-free DNA in plasma of patients. (F) IFN-γ ELISPOT assay for T cells reactive against select tumor antigens, VSV-N, or cytomegalovirus, Epstein-Barr virus, and Flu virus positive control. (G) Anti-VSV neutralizing antibody titers (average of patients per DL). Mean ± standard error of the mean shown where applicable.
Pharmacodynamics of VSV infection can be monitored by measuring the profile of virally encoded IFN-β in the plasma over time (Figure 4C). Peak IFN-β was notably higher in patients who achieved a clinical response to treatment vs those who did not (median, 662.6 vs 205.3; P = .18). This difference did not reach statistical significance, but given the limited numbers of patients, these are exploratory findings that can be used in further studies. In patient 4A.3 (CR) and patient 4A.4 (PR) peak IFN-β levels were 18 213 and 662 pg/mL, respectively. Patients with TCL tended to have higher peak IFN-β (Figure 4D). In fact, higher DL was positively and significantly associated with higher attained peak IFN-β levels (r = 0.79; P = .0005). There was also a significant positive correlation (r = 0.64; P = .012) between peak IFN-β achieved and peak viremia levels, emphasizing the importance of achieving a high enough dose for virus delivery and infection of tumors.
VSV has a fast replication cycle and rapidly induces cell lysis. Indeed, plasma cell-free DNA increased significantly from baseline in patients 4A.3 and 4A.6 with robust viral replication, as indicated by the high plasma IFN-β at 18 213 and 2650 pg/mL, respectively (Figure 4E). Patient 4A.5 with minimal viral infection, reflected in the very low (17 pg/mL) IFN-β at 24 hours, showed no change in plasma cell-free DNA. To investigate if VSV virotherapy and cell lysis might have subsequently increased immune responses to tumor antigens, peripheral blood mononuclear cell (PBMC) from patients with clinical responses were incubated with peptide pools against a panel of tumor-associated antigens (NYESO1, WT1, PRAME, MAGE A1, MAGE A3, MAGE C1, hTERT, Muc-1, p53) to VSV-N and cytomegalovirus, Epstein-Barr virus, and Flu virus as positive control. T-cell responses against VSV-N increased in all patients, most significantly in patients 4A.3 (CR) and 4A.4 (PR). Patient 2A.1 (PR) had strong baseline T-cell responses against several tumor antigens, which did not increase further after VSV. Increases in tumor antigen reactive T cells were seen in patients 4A.3 and 4A.4 after VSV (Figure 4F). In contrast, patient 4A.1, who did not have a clinical response, did not show boosting of tumor antigen-reactive T cells.
Discussion
We have demonstrated the safe IV administration of OV VSV-IFNβ-NIS at doses up to 1.7 × 1011 TCID50 in patients with relapsed and refractory hematologic malignancies. Remarkable antitumor activity was documented, most notably at the highest DL (DL4) and in patients with TCL.
One short-lived PR lasting 3 months was observed in a patient with TCL at DL2. It was at DL4 that substantial responses emerged: an ongoing CR 20 months after VSV and a PR lasting 6 months in patients with TCL and the first observation of disease stability in patients with myeloma. This dose-response relationship had been observed in our preclinical work in mice bearing 5TGM1 murine plasmacytomas, in which tumor eradiation was only seen with the highest tolerated dose of VSV-mIFNβ-NIS.17,20 The clinical responses seen at DL4 underscore that for IV administration of OV, and dose is critical to achieve the required viral concentration for adequate seeding of intratumoral virus foci at a sufficient density to mediate tumor destruction.30 A dose threshold for intratumoral delivery by a systemically administered vaccinia virus was also previously reported.31 The variable responsiveness of different tumor histologies at a given DL seen in this trial is in keeping with preclinical studies showing that the absolute dose of virus needed to impact tumor growth varies greatly from one tumor model to another. Factors such as the intrinsic susceptibility of tumor cells to virus propagation and the permeability of tumor neovessels to circulating virus particles contribute to this variability.9,11,32
Interestingly, whereas durable responses were seen in 2 of the 4 patients with TCL treated at DL4, lesser responses, including short-lived PR and/or mixed responses, were observed in 3 additional patients with TCL at DL2 and 3. The mixed responses (supplemental Tables 1-2), in which some lesions regressed while others were unaffected, suggest that permissiveness to virus extravasation and/or intratumoral amplification can vary significantly even from 1 tumor deposit to another in each patient with cancer. VSV enters cells via the low-density lipoprotein receptor and is endocytosed, where upon acidification of the endosomes, the viral nucleocapsid is released into the cytoplasm for transcription and translation of viral genes and genomes.33 Variability in low-density lipoprotein receptor expression on tumor cells, constitutive expression of antiviral genes due to activated JAK/STAT pathways in TCL, and overactive protein kinase R-like endoplasmic reticulum kinase, which phosphorylates eIF2α, reducing protein synthesis could contribute to differences in permissiveness to VSV replication and oncolysis among patients.33-36
Aside from OV dose, the absence of antiviral antibodies was likely a key factor contributing to the success of the therapy in this study. None of the trial subjects had detectable anti-VSV antibodies at baseline, but most had detectable rising antibody titers by day 8 after therapy. Of relevance to this observation, our preclinical studies have shown that mice bearing VSV-responsive tumors become completely resistant to oncolytic VSV therapy after passive transfer of VSV-immune serum.37 Overall, these findings suggest that there may be a short window of opportunity for administration of a second IV virus infusion, which could be tested as a future dose escalation strategy.
IV VSV-IFNβ-NIS was relatively well tolerated in the current study at all 4 DLs, and dose-limiting toxicities were prominent at the highest DL, with 83% of patients (5/6) experiencing transient CRS. The CRS events responded well to fluid bolus administration, except in 1 patient treated at DL2, who required transient single vasopressor support over 4 hours. Premedication with Tylenol, nonsteroidal antiinflammatory drugs, antiemetics, and IV fluids was subsequently implemented for CRS prevention, but even with these interventions, 5 of 6 patients treated at DL4 did develop grade 2 CRS. Future studies will evaluate the protective effect of prophylactic dexamethasone therapy.
Hematologic AEs, particularly lymphopenia, were the prominent grade 3 and 4 AEs. The protocol initially included patients with AML, but the preexisting cytopenias in the patient with AML treated at DL2 became more profound after virus infusion, manifesting as grade 3 anemia and grade 4 neutropenia, thrombocytopenia, and lymphopenia. The recruitment strategy thereafter refocused on patients with TCL and MM. Overall, these clinical findings showcase the manageable AE profile with systemic VSV-IFNβ-NIS, which may even challenge the safety profiles of other novel therapies such as chimeric antigen receptor T cell (CAR-T) platforms for myeloma or mogamulizumab for cutaneous TCL.38-40
VSV-IFNβ-NIS was designed to facilitate the noninvasive in vivo tracking of intratumoral virus replication in treated patients, either by NIS reporter gene imaging using monovalent radioiodine, pertechnetate, or tetrafluoroborate anions19 or by serial determination of plasma levels of virally encoded IFN-β. However, in this study, with exception of 1 patient who had multiple plasmacytomas, 99mTc-pertechnetate SPECT/CT did not provide clear images of virus infection, even among the responding patients. NIS imaging could be improved by using PET tracers, which offer 2 to 3 orders of magnitude increase in sensitivity of detection.41,42 Therefore, we are transitioning to using the more sensitive 18F-tetrafluoroborate PET tracer for NIS imaging in our amended protocol.43,44
Analysis of the plasma IFN-β concentration profiles of virus-treated patients confirms that serum level of virus-encoded IFN-β can serve as a convenient biomarker for intratumoral virus amplification. Analyses for multiple cytokines other than IFN-β showed peak plasma concentrations at the 4-hour timepoint after virus infusion, returning to baseline by 24 hours. In contrast, in several study subjects, the serum level of IFN-β continued to rise beyond the 4-hour postinfusion timepoint, in some cases to very high levels with persistence for several days. Given the distinct and prolonged serum concentration profiles observed for IFN-β and the high patient-to-patient variability in peak serum levels at each DL, we conclude that the IFN-β must be derived in large part from VSV-IFNβ-NIS–infected tumor cells. This is in keeping with the results of our preclinical studies in tumor-bearing mice and dogs, in which intratumoral virus spread of VSV-IFNβ-NIS is accompanied by the release of virus-encoded IFN-β into the bloodstream and peak serum IFN-β levels can be used to track the extent of the virus infection.17,21,45
Peak serum levels of IFN-β in the current clinical trial were typically seen at 24 hours after therapy, which suggests that the infused VSV-IFNβ-NIS virus induces only a short-lived intratumoral infection.
However, the magnitude of IFN-β production varied greatly among study subjects, with peak concentrations at 24 hours ranging from a low of 17 pg/mL in a patient with a completely nonresponsive tumor to a high of 18 213 pg/mL in the patient who achieved a CR. In keeping with our preclinical studies elucidating the potential toxicities of virally encoded IFN-β, higher IFN-β levels in the current study were associated with transient elevations of alanine aminotransferase and aspartate aminotransferase as well as reversible thrombocytopenia. Interestingly, not all patients generating a high IFN-β level responded well to the therapy, indicating that it is possible to have a relatively extensive virus infection in the tumor without necessarily seeing an associated clinical response. In addition, type I interferon (IFN-α) has antitumor activity against cutaneous TCL and could certainly add to the activity of VSV-IFNβ-NIS in the patients.46 The complex relationship among oncolytic killing, immune killing, and tumor regression after infusion of VSV-IFNβ-NIS is clearly an important topic in need of elucidation in larger clinical studies.
Although the overall number of subjects enrolled in this phase 1 study is small, the efficacy signal in TCL is remarkable. Despite several recent advances, such as the advent of brentuximab vedotin for CD30+ TCL, the prognosis for this disease remains guarded with few upfront treatment options outside classic anthracycline-based regimens, especially for PTCL, and survivals estimated from 2 to 15 months for those with relapsed/refractory disease.40,47 In the current study, half of the patients with TCL treated with VSV-IFNβ-NIS at DL4 responded. All the patients enrolled had already exhausted other treatment options and were attracted to the study in part because VSV-IFNβ-NIS requires only a single administration. Among the MM cohort, best response of stable disease suggests initial activity with VSV-IFNβ-NIS but indicates that a combination of chemo-immunomodulatory agents may be required to engage a deeper and more sustained response.
In summary, we have shown that a single IV administration of VSV-IFNβ-NIS is safe up to a dose of 1.7 × 1011 TCID50 and demonstrates compelling dose-dependent efficacy among patients with advanced treatment-refractory TCL. We are currently enrolling an expansion cohort of additional patients with relapsed, refractory PTCL at DL4. Additional arms have also been added to the ongoing phase 1 trial to investigate the safety and efficacy of using VSV-IFNβ-NIS in combination with drugs that modulate either antiviral or antitumor immune responses. As immune-directed strategies continue to redefine the approach to cancer treatment, the clinical potential of OV with VSV-IFNβ-NIS appears to be another safe and promising modality in the therapeutic armamentarium for relapsed hematologic malignancies.
Acknowledgments
The authors thank research technologists Ms. Alysha Newsom and Mr. Michael Steele and sincerely thank the patients who participated in the trial. The Mayo Clinic Viral Vector Production Laboratory was responsible for manufacture of the cGMP virus.
This study was funded by the National Cancer Institute, National Institutes of Health, Mayo Clinic Multiple Myeloma SPORE (grant P50CA186781), the David F. and Margaret T. Grohne Foundation, the Lu Family Foundation, and the Philips Foundation.
Authorship
Contribution: M.Q.L., S.J.R., T.E.W., M.P., A.C.D., S.G., and K.-W.P. designed the research; J.C., K.-W.P., T.E.W., S.B., J.C.V., J.P., M.P., V.R., A.D., N.L., F.B., N.B., S.M.A., L.Z., N.P., B. Balakrishnan, B. Brunton, M.G., B.G., A.C.D., M.A.G., R.W., R.S.G., S.R.H., D.D., S.K., L.B., W.G., T.K., E.M., P.K., R.A.K., Y.L., S.N., J.L.M., M.S., A.F., M.H., L.H., S.J.R., and M.Q.L. performed research and collected and analyzed data; S.G., A.C.D., and B.G. performed statistical analysis; and J.C., M.Q.L., K.-W.P., and S.J.R. wrote the manuscript.
Conflict-of-interest disclosure: S.J.R., K.-W.P., and S.N. own equity in Vyriad, an oncolytic virotherapy company. J.L.M. served as a consultant for Pharmacyclics/AbbVie, Bayer, Gilead/Kite Pharma, Pfizer, Janssen, Juno/Celgene, BMS, Kyowa, Alexion, Fosunkite, Innovent, Seattle Genetics, Debiopharm, Karyopharm, Genmab, ADC Therapeutics, Epizyme, Beigene, Servier, Novartis, Morphosys/Incyte, and Lilly; received research funding from Bayer, Gilead/Kite Pharma, Celgene, Merck, Portola, Incyte, Genentech, Pharmacyclics, Seattle Genetics, Janssen, and Millennium; received honoraria from Targeted Oncology, OncView, Curio, Kyowa, Physicians' Education Resource, and Seattle Genetics; and served on the speaker’s bureau of Gilead/Kite Pharma, Kyowa, Bayer, Pharmacyclics/Janssen, Seattle Genetics, Acrotech/Aurobindo, Beigene, Verastem, AstraZeneca, Celgene/BMS, and Genentech/Roche. N.B. served on the advisory board of Verastem, Purdue Pharma, Daichii Sankyo, Kyowa Kirin, Vividion, Kymera, and Secura Bio and was an invited speaker for Medscape. The remaining authors declare no competing financial interests.
Correspondence: Martha Q. Lacy, Division of Hematology, Mayo Clinic, 200 First St SW, Rochester, MN 55905; e-mail: lacy.martha@mayo.edu.

