

Key Points
Rare copy number variants in FAS can cause ALPS by method of haploinsufficiency.
CNV analysis can provide a molecular diagnosis for patients with ALPS when whole exome sequencing or panel-based testing are inconclusive.
Abstract
Autoimmune lymphoproliferative syndrome (ALPS) is characterized by chronic nonmalignant lymphadenopathy, splenomegaly, cytopenias, and other autoimmune manifestations. ALPS is caused by lymphocyte accumulation from defects in FAS-mediated apoptosis. Heterozygous germline or somatic pathogenic single nucleotide variants in FAS are the most common molecular etiology of ALPS. Through the Centralized Sequencing Program at the National Institute of Allergy and Infectious Diseases, we performed exome sequencing on subjects with a clinical diagnosis of ALPS, with a subset receiving copy number variant (CNV) analysis. In this cohort, we identified 3 subjects from unrelated families with CNVs at the FAS locus. One subject had a de novo ∼0.828 Mb copy number loss encompassing all of FAS. The second subject had a maternally inherited ∼1.004 Mb copy number loss encompassing all of FAS. The third subject had a paternally inherited ∼0.044 Mb copy number loss encompassing exons 7 through 9 of FAS. Subjects with deletions in FAS had clinical presentations and biomarker profiles similar to those with ALPS and with germline and somatic FAS variants. We demonstrate that CNV analysis should be pursued if there is clinical and biomarker evidence of ALPS because it can lead to a molecular diagnosis and appropriate treatment when FAS sequencing is inconclusive.
Introduction
Autoimmune lymphoproliferative syndrome (ALPS) is a disorder of immune dysregulation occurring from a disruption in the FAS-mediated lymphocyte apoptosis pathway, resulting in chronic nonmalignant lymphadenopathy, splenomegaly, cytopenias, and other autoimmune manifestations.1,2 Variants affecting FAS homotrimer formation, especially those in the highly conserved FAS death domain, contribute to a dominant-negative effect on apoptosis through disruption of FAS receptor assembly.2 In contrast, variants causing premature terminations have been reported to contribute to ALPS via haploinsufficiency.2 Reduced FAS levels result in decreased binding of FAS-L, leading to diminished apoptosis.2
Heterozygous germline and somatic pathogenic FAS variants account for 70% and 20% of all ALPS diagnoses, respectively.3 Fewer than 1% of cases result from large FAS deletions or intronic FAS variants that lead to exonic deletions.3 Homozygous or compound heterozygous variants in FAS typically result in high penetrance and severe, early onset of disease, whereas heterozygous variants are less penetrant.4-7 Furthermore, missense variants that affect the intracellular domain are more penetrant (63%-90%) compared with missense variants that affect the extracellular domain (30%-52%).4 Loss-of-function variants resulting in haploinsufficiency are also characterized by incomplete penetrance and variable expressivity.8
To our knowledge, there are only 4 cases of deletions in FAS that are reported to cause ALPS.9 A 290-bp deletion resulting in a smaller transcript and addition of a 7 amino-acid peptide extending into the death domain was reported to cause ALPS in an individual with hepatosplenomegaly, lymphocytosis, lymphadenopathy, autoimmune hemolytic anemia, and neutropenia.5 A germline 5′ splice-site SNV in the third intron of FAS caused deletion of the entire third exon in an individual presenting with lymphadenopathy, hepatosplenomegaly, neutropenia, hemolytic anemia, elevated interleukin-10, and elevated double-negative T (DNT) cells.10 Moreover, a paternally inherited germline deletion that included exon 6 of FAS as well as a somatic missense mutation (p.Ala237Pro) located in exon 9 (the death domain) of FAS were reported in an individual with splenomegaly, lymphadenopathy, neutropenia, immune thrombocytopenia, autoimmune hemolytic anemia, and the presence of antinuclear antibodies.11 In the DNT cells, lower FAS expression was reported in this individual relative to his father. Consistent with reduced penetrance observed in ALPS, the father was clinically asymptomatic, but expressed lower FAS than controls by flow cytometric analysis.8,11 In addition, a 28-bp deletion c.985_1008+4, encompassing a canonical splice site, was reported to potentially cause haploinsufficiency in an individual with ALPS.2 This deletion occurred after the death domain and is potentially analogous to a c.987ins20 frameshift variant reported in a patient with undetectable surface expression of FAS.2 Finally, a loss of function IVS3 as A-G −2 homozygous splice variant that led to skipping of exon 4 and complete lack of FAS receptor expression was reported in a consanguineous patient with lymphadenopathy, hepatosplenomegaly, pancytopenia, hypergammaglobulinemia, and increased DNT cells.7
The clinical significance of deletions in ALPS undetectable by exome sequencing and their relatively rare occurrence prompt the need for further investigation into the phenotypic spectrum of patients with copy number losses in FAS, as well as the consideration of copy number variant (CNV) testing in the context of ALPS. Here, we describe 3 unrelated families with unique large deletions at the FAS locus underlying ALPS. Two subjects harbored deletions encompassing the entire gene and 1 subject harbored a deletion that encompassed exons 7 through 9. This case series presents additional clinical data and showcases the importance of CNV analyses when there is significant clinical and biomarker evidence consistent with ALPS in the absence of somatic or germline FAS variants.
Patients and methods
All patients were enrolled and evaluated at the National Institutes of Health through institutional review board-approved protocol (#NCT00001350). Subjects gave informed consent and underwent genomic workup to identify molecular contributions to immunological disorders.
Exome sequencing on genomic DNA was performed on an Illumina platform. The exonic regions, flanking splice junctions, and 5′ and 3′ untranslated regions were sequenced with 100-bp or greater paired-end reads with a minimum coverage of 95% >20×. Reads were aligned to GRCh37/UCSC hg19 and analysis was performed on the Genomic Research Integration System (gris.niaid.nih.gov), with the custom-enhanced tool GRIS-SEQR.12 Any relevant variants were validated by Sanger sequencing in a Clinical Laboratory Improvement Amendments/College of American Pathologists laboratory.
Chromosomal microarray analysis (CMA) was also performed on a 180 000-probe custom-designed Agilent oligonucleotide microarray (NIAID chip, v1), which targets ∼2400 genes potentially involved in human immunity at single-exon resolution. CNVs were detected and reported based on their locations and frequency in the Baylor Genetics internal database. Genomic linear positions were reported relative to GRCh37/UCSC hg19.
Results and discussion
Exome sequencing was completed on 94 subjects with a clinical diagnosis suggestive of ALPS. For 33 subjects without a molecular diagnosis from exome, we performed CMA analysis. We identified 3 subjects with a copy number loss involving FAS (Table 1; Figure 1). All 3 individuals manifested ALPS primary criteria in childhood. Two individuals exhibited defective apoptosis. All 3 exhibited several secondary criteria: hemolytic anemia, thrombocytopenia, neutropenia, elevated B12 levels, elevated immunoglobulin G, and had negative family histories of lymphoma and lymphoproliferation (Table 1). Parental studies were performed for all families. Parental studies did not detect the copy number loss in subject 1’s parents, suggesting that this change arose de novo in this subject. In family 2, the copy number loss was shown to be inherited from an apparently unaffected mother. Furthermore, the unaffected siblings were not tested. In family 3, parental studies showed the copy number loss to be paternal. Additionally, the unaffected sister did not harbor the copy number loss (Table 1; supplemental Figure 1).
CNV results and clinical phenotypes of probands with autoimmune lymphoproliferative syndrome
| Subject 1 (140.1) | Subject 2 (384.1) | Subject 3 (396.1) | |
|---|---|---|---|
| CNV results | |||
| Size of copy number loss, Mb | 0.828 | 1.004 | 0.044 |
| Exons affected | All | All | 7-9 |
| Genomic location | 10q23.31 | 10q23.31 | 10q23.31 |
| Probes, no. | 15 | 63 | 15 |
| Parental studies | De novo copy number loss | Maternally inherited, mother with mild neutropenia and no other manifestations | Paternally inherited, father unaffected |
| Clinical phenotypes | |||
| Age, y | 34 | 11 | 14 |
| Race | Caucasian | Asian | Caucasian |
| Age of onset, y | 5 | 2 | 5 |
| Presenting symptoms | Arthralgias, autoimmune fevers, diarrhea, and autoimmune hemolytic anemia, adenopathy, splenomegaly, pancytopenia, and follicular hyperplasia | Severe pancytopenia, hepatosplenomegaly, and lymphadenopathy | Fatigue, abdominal pain, cytopenias, lymphadenopathy, and splenomegaly |
| Treatment | Prednisone pulses No sirolimus No mycophenolate | Mycophenolate | Mycophenolate with poor response, sirolimus with good effect |
| Required criteria | |||
| Splenomegaly | + | + | + |
| Lymphadenopathy | + | + | + |
| Elevated CD3+ αβ+ CD4−CD8− DNT cells (>1.5% of total lymphocytes or >2.5% of CD3+ lymphocytes) with normal or elevated lymphocyte counts | + | + | + |
| Accessory criteria | |||
| Primary criteria | |||
| Defective FAS-induced apoptosis | + (22.2%; absolute of 325) | NR (9.25%; no absolute provided) | + |
| Somatic or germline pathogenic mutation in FAS, FASL, FADD, or CASP10 | – | – | – |
| Secondary criteria | |||
| Hemolytic anemia | + | + | + |
| Thrombocytopenia | + | + | + |
| Neutropenia | + | + | + |
| Serum elevated B12 levels (>1500 pg/mL) | + | + | + |
| Elevated plasma sFASL levels (>200 pg/mL) | – | + (2193 pg/mL) | – |
| Elevated plasma interleukin-10 levels (>20 pg/mL) | NR | + (27 pg/mL) | + |
| Typical immunohistopathology findings (paracortical T-cell hyperplasia) | – | + | – |
| Elevated immunoglobulin G levels (polyclonal hypergammaglobinemia) | + | + | + |
| Family history of a nonmalignant or lymphoma-associated noninfectious lymphoproliferation with or without autoimmunity | – | – | – |
| Subject 1 (140.1) | Subject 2 (384.1) | Subject 3 (396.1) | |
|---|---|---|---|
| CNV results | |||
| Size of copy number loss, Mb | 0.828 | 1.004 | 0.044 |
| Exons affected | All | All | 7-9 |
| Genomic location | 10q23.31 | 10q23.31 | 10q23.31 |
| Probes, no. | 15 | 63 | 15 |
| Parental studies | De novo copy number loss | Maternally inherited, mother with mild neutropenia and no other manifestations | Paternally inherited, father unaffected |
| Clinical phenotypes | |||
| Age, y | 34 | 11 | 14 |
| Race | Caucasian | Asian | Caucasian |
| Age of onset, y | 5 | 2 | 5 |
| Presenting symptoms | Arthralgias, autoimmune fevers, diarrhea, and autoimmune hemolytic anemia, adenopathy, splenomegaly, pancytopenia, and follicular hyperplasia | Severe pancytopenia, hepatosplenomegaly, and lymphadenopathy | Fatigue, abdominal pain, cytopenias, lymphadenopathy, and splenomegaly |
| Treatment | Prednisone pulses No sirolimus No mycophenolate | Mycophenolate | Mycophenolate with poor response, sirolimus with good effect |
| Required criteria | |||
| Splenomegaly | + | + | + |
| Lymphadenopathy | + | + | + |
| Elevated CD3+ αβ+ CD4−CD8− DNT cells (>1.5% of total lymphocytes or >2.5% of CD3+ lymphocytes) with normal or elevated lymphocyte counts | + | + | + |
| Accessory criteria | |||
| Primary criteria | |||
| Defective FAS-induced apoptosis | + (22.2%; absolute of 325) | NR (9.25%; no absolute provided) | + |
| Somatic or germline pathogenic mutation in FAS, FASL, FADD, or CASP10 | – | – | – |
| Secondary criteria | |||
| Hemolytic anemia | + | + | + |
| Thrombocytopenia | + | + | + |
| Neutropenia | + | + | + |
| Serum elevated B12 levels (>1500 pg/mL) | + | + | + |
| Elevated plasma sFASL levels (>200 pg/mL) | – | + (2193 pg/mL) | – |
| Elevated plasma interleukin-10 levels (>20 pg/mL) | NR | + (27 pg/mL) | + |
| Typical immunohistopathology findings (paracortical T-cell hyperplasia) | – | + | – |
| Elevated immunoglobulin G levels (polyclonal hypergammaglobinemia) | + | + | + |
| Family history of a nonmalignant or lymphoma-associated noninfectious lymphoproliferation with or without autoimmunity | – | – | – |
NR, not reported.
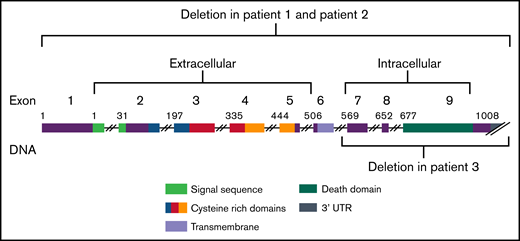
FAS gene regions. Graphic of FAS deletions showing the copy number variants identified here in autoimmune lymphoproliferative syndrome.
FAS gene regions. Graphic of FAS deletions showing the copy number variants identified here in autoimmune lymphoproliferative syndrome.
We performed exome sequencing in individuals with a clinical diagnosis of ALPS, and CMA for those without molecular diagnoses from exome. We found 3 cases of CNVs underlying the clinical diagnosis. Our results are consistent with the pathogenic nature of CNV losses involving FAS. The FAS deletions we identified encompassed the whole gene in 2 subjects and exons 7 through 9 in 1. All subjects exhibited clinical manifestations and biomarker evidence that was indistinguishable from individuals with ALPS caused by pathogenic germline or somatic SNVs. The mechanism of disease in these subjects was consistent with haploinsufficiency because each subject still had 1 functional copy of FAS.
The phenotypes of the parents harboring the FAS deletion, including the mother of subject 2 who was asymptomatic with only a history of mild neutropenia, and the father of subject 3, who was unaffected, are illustrative of the reduced penetrance that is seen in families with ALPS.1 This raises the possibility that genetic modifiers or allele-specific expression may contribute to the disease phenotype.
Our results demonstrate that, when germline or somatic FAS SNVs are not found, CNV analysis should be performed. Identification of CNVs in FAS would both allow for better management and treatment recommendations, such as monitoring for lymphoma.13 Additionally, individuals with FAS CNVs could potentially benefit from gene therapy, as previously suggested for haploinsufficient somatic SNV cases showing that increasing FAS surface expression can increase apoptosis levels to that of controls.8
This report has several limitations. First, more individuals with FAS CNVs need to be studied to establish genotype-phenotype correlations. Second, these subjects were ascertained in a clinical cohort of individuals meeting ALPS criteria. There may be patients harboring FAS deletions who do not meet the same criteria; further assessment of the clinical spectrum is warranted. Finally, our argument that pathogenic FAS CNVs cause haploinsufficient ALPS with reduced penetrance would be strengthened by pathway analysis, functional studies, and whole-genome sequencing.
In summary, this report showcases the benefit of performing CMA when panel-based testing or exome sequencing for ALPS are inconclusive. The clinical phenotypes and familial segregation analyses support haploinsufficiency as a mechanism of disease and the autosomal dominant inheritance model with reduced penetrance seen in ALPS resulting from FAS defects.
Acknowledgments
The authors thank the patients and their families for participating in our study and providing samples. They also thank the Department of Laboratory Medicine team, Yan Su, Julie Niemela, and Sujin Hwang for processing and preparing the samples; the Bioinformatics and Computational Biosciences Branch team, Justin Lack and Vasu Kuram, for providing bioinformatic support; and the Genomic Research Integration System team for their assistance in this study. They also thank Amy Breman, Weimin Bi, and Pawel Stankiewicz for assistance on the design of the custom copy-number microarray.
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health (75N91019D00024). This research was funded by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases at the National Institutes of Health Clinical Center. L.M.F. is supported by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases at the National Institutes of Health. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Authorship
Contribution: K.J., S.P., M. Similuk, E.K., J.Y., M. Setzer, L.J., L.M.F., R.G., M.W., and V.K.R. all made substantial contributions to the work, as outlined by the International Committee of Medical Journal Editors, including the conception or design of the work and contributing to drafting and revising the work critically for important intellectual content; K.J. collected data for the study and drafted the manuscript; K.J., S.P., E.K., and V.K.R. reviewed the clinical data; S.P., E.K., M. Similuk, M. Setzer, L.J., L.M.F., and V.K.R. made substantial contributions to the clinical care and evaluation of these patients; L.M.F. designed the copy-number microarray; M.W. and R.G. analyzed the genomic data and made substantial contributions to its interpretation; E.K., J.Y., M.W., and V.K.R. contributed to drafting the work and revising it critically for important intellectual content; and all authors provided final approval of the version to be published, and agree to be accountable for all aspects of the work.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kathleen Jevtich, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Room 11C207 Building 10, 10 Center Dr, Bethesda, MD 20814; e-mail: katy.jevtich@gmail.com.

