

Key Points
GRK2 regulates the hemostatic response by limiting ADP P2Y1- and P2Y12-mediated signaling.
Maintaining GRK2 activity in platelets may be beneficial for prevention of thrombotic diseases.

Abstract
The critical role of G protein–coupled receptor kinase 2 (GRK2) in regulating cardiac function has been well documented for >3 decades. Targeting GRK2 has therefore been extensively studied as a novel approach to treating cardiovascular disease. However, little is known about its role in hemostasis and thrombosis. We provide here the first evidence that GRK2 limits platelet activation and regulates the hemostatic response to injury. Deletion of GRK2 in mouse platelets causes increased platelet accumulation after laser-induced injury in the cremaster muscle arterioles, shortens tail bleeding time, and enhances thrombosis in adenosine 5′-diphosphate (ADP)-induced pulmonary thromboembolism and in FeCl3-induced carotid injury. GRK2−/− platelets have increased integrin activation, P-selectin exposure, and platelet aggregation in response to ADP stimulation. Furthermore, GRK2−/− platelets retain the ability to aggregate in response to ADP restimulation, indicating that GRK2 contributes to ADP receptor desensitization. Underlying these changes in GRK2−/− platelets is an increase in Ca2+ mobilization, RAS-related protein 1 activation, and Akt phosphorylation stimulated by ADP, as well as an attenuated rise of cyclic adenosine monophosphate levels in response to ADP in the presence of prostaglandin I2. P2Y12 antagonist treatment eliminates the phenotypic difference in platelet accumulation between wild-type and GRK2−/− mice at the site of injury. Pharmacologic inhibition of GRK2 activity in human platelets increases platelet activation in response to ADP. Finally, we show that GRK2 binds to endogenous Gβγ subunits during platelet activation. Collectively, these results show that GRK2 regulates ADP signaling via P2Y1 and P2Y12, interacts with Gβγ, and functions as a signaling hub in platelets for modulating the hemostatic response to injury.
Introduction
Blood platelets are small, anucleate cells that play a central role in hemostasis, thrombosis, and inflammation. Prior to injury, platelets circulate in a discoid resting state. When challenged by vascular injury, platelets rapidly activate and aggregate with each other, forming a plug on the vessel wall to prevent vascular leakage. Platelet activation needs to be regulated so that platelets can be prepared for rapid activation after vascular injury to prevent bleeding while at the same time avoiding an overly exuberant response to injury that may lead to ischemia, heart attack, or stroke. Although initial adhesion of platelets to the damaged vessel wall is driven by collagen, subsequent recruitment of additional platelets into a growing thrombus requires mediators such as thrombin, thromboxane A2 (TxA2), and adenosine 5′-diphosphate (ADP), all of which act through G protein–coupled receptors (GPCRs).1,2 Mechanisms that provide negative feedback toward activated GPCRs to limit platelet activation and thrombus formation are thus key to achieving an optimal response to vascular injury. Emerging studies from our group and others strongly suggest that GPCR kinases (GRKs) are critical negative regulators for platelet activation.3-7
GRKs are a group of serine/threonine kinases that typically terminate agonist-occupied GPCR signaling in a phosphorylation-dependent manner. In cells other than platelets, GRKs phosphorylate agonist-bound GPCRs, triggering arrestin binding followed by clathrin-mediated endocytosis.8 We have been able to detect the expression of GRK2, GRK5, and GRK6 in platelets.3 Over the last 30 years, the role of GRK2 in cardiovascular disease has been widely studied, and it has been reported that GRK2 messenger RNA and its activity increase in patients with cardiac ischemia and hypertrophy.9,10 Conversely, the impact of GRK2 on hemostasis and thrombosis remains unknown. Using a transfected cell assay, Hardy et al11 suggest that GRK2 is involved in ADP P2Y12 receptor–mediated platelet desensitization. However, little is known about the negative feedback mechanisms governing ADP P2Y receptor–mediated platelet activation.
Here, we show that GRK2−/− mice display greater platelet activation and platelet accumulation at the site of the injury. The increased responsiveness to agonists in GRK2−/− platelets is limited to ADP P2Y1- and P2Y12-mediated signaling. Absence of GRK2 in platelets increases thrombosis in ADP-induced pulmonary thromboembolism and in FeCl3-induced carotid injury. P2Y12 antagonist treatment eliminates the phenotypic difference in platelet accumulation between wild-type (WT) and GRK2−/− mice in response to injury. Pharmacologic inhibition of GRK2 in human platelets causes an increase in platelet activation in response to 2-methylthioadenosine diphosphate (2MesADP). Collectively, these observations show for the first time that GRK2 functions as a critical negative regulator in hemostasis and thrombosis.
Methods
Vascular injury: platelet accumulation and fibrin generation
GRK2−/− (GRK2fl/fl/Pf4-Cre+) male mice with matched WT control littermates (GRK2fl/fl/Pf4-Cre-) aged 8 to 12 weeks were used. Briefly, Alexa Fluor 568–labeled anti-CD41 antibody F(ab)2 fragments, Alexa Fluor 488–labeled anti–P-selectin, and Alexa Fluor 647–labeled fibrinogen were administered via the retro-orbital plexus. Arterioles in the cremaster muscle of 30 to 50 μm in diameter were studied. Vascular injury was induced by using a laser ablation system (Ablate! photoablation system; Intelligent Imaging Innovations [3i]). Thrombus formation was observed for 3 minutes and analyzed as previously described.3 We also examined laser-induced thrombus formation in WT and GRK2−/− mice treated with a P2Y12 antagonist, cangrelor. Cangrelor (0.75 μg before each injury) or vehicle (saline) was administered via IV infusion through the jugular vein.
The studies involved in the research reported in this article have been approved by the Thomas Jefferson University Institutional Review Board and Institutional Animal Care and Use Committee. The study was conducted in accordance with the Declaration of Helsinki.
Pulmonary thromboembolism
Pulmonary thromboembolism assay was performed as we previously described.6 Briefly, Alexa Fluor 750–labeled anti-GPIX was administered IV into anesthetized WT and GRK2−/− mice. After 20 minutes, the anesthetized mice were dosed with ADP (0.3 mg/g) IV. The lung was imaged by using the Licor imaging system.
Statistical analysis
Results are presented as mean ± standard error of the mean. Data were analyzed by using the t test or analysis of variance followed by appropriate post hoc test. P < .05 was considered statistically significant. Additional statistical analysis and methods are described in the supplemental Figures 3, 4, 8, 9, and 10 and Figure legends.
Results
Generation and characterization of GRK2−/− mice
GRK2−/− mice die during gestation due to retardation of cardiac development.12,13 Thus, we took advantage of a GRK2 conditional knockout mouse model. We crossed “floxed” GRK2 mice, containing two loxP sites in intron 2 and intron 6 of the GRK2 locus,13 with Pf4-Cre mice expressing the Cre recombinase under the control of the megakaryocyte-specific platelet factor-4 (Pf4) promoter. This allowed for deletion of GRK2 specifically in megakaryocytes and platelets (Figure 1A; supplemental Figure 1).14 GRK2flox/flox/Pf4-Cre– are henceforth referred to as WT, and GRK2flox/flox/Pf4-Cre+ are further referred to as GRK2−/− in this study.
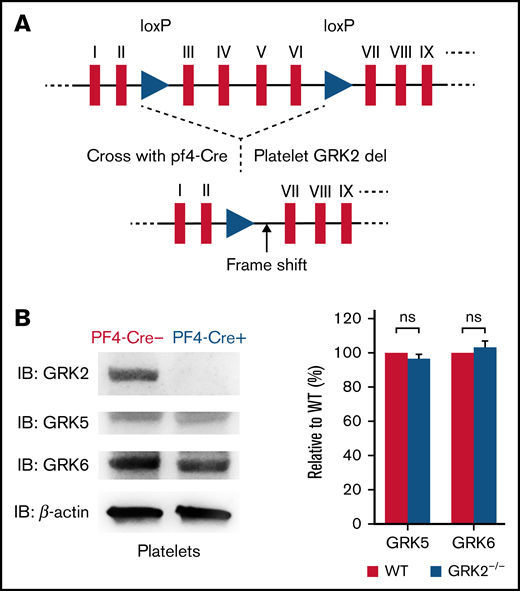
Generation and characterization of platelet-specific deletion of GRK2 in mouse. (A) Schematic map of loxP sites inserted in the mouse GRK2 gene. Two loxP sites flanking exons III through VI of mouse GRK2 are shown as triangles. (B) Western blot images of platelet lysate from PF4-Cre– and PF4-Cre+ mice probed with the GRK2-, GRK5-, and GRK6-specific antibodies. Deletion of GRK2 in platelets does not affect the expression of GRK5 and GRK6. n = 4. Supplemental Figure 2 features the full blots. ns, not significant.
Generation and characterization of platelet-specific deletion of GRK2 in mouse. (A) Schematic map of loxP sites inserted in the mouse GRK2 gene. Two loxP sites flanking exons III through VI of mouse GRK2 are shown as triangles. (B) Western blot images of platelet lysate from PF4-Cre– and PF4-Cre+ mice probed with the GRK2-, GRK5-, and GRK6-specific antibodies. Deletion of GRK2 in platelets does not affect the expression of GRK5 and GRK6. n = 4. Supplemental Figure 2 features the full blots. ns, not significant.
GRK2−/− mice grew and developed normally. This platelet-specific deletion of GRK2 in mice did not affect the expression of the other 2 major GRKs (GRK5 and GRK6) in platelets (Figure 1B; supplemental Figure 2A). The platelet number, mean platelet volume, red blood cell count, and white blood cell count of GRK2−/− mice were comparable to those of littermate WT control mice (supplemental Table 1). The surface expression of integrin is normal on GRK2−/− platelets (supplemental Figure 2B).
GRK2−/− mice display increased platelet accumulation at the outer layer of the thrombus in response to injury
Platelet function in vivo was assessed with real-time confocal fluorescence microscopy following a laser-induced injury in the cremaster muscle arterioles.15 Hemostatic thrombi that form after penetrating injuries in this setting have a characteristic core/shell architecture in which the extent of platelet activation is determined by the distribution and concentration of agonists in the immediate environment of each platelet. In the core region of the thrombus, thrombin mediates platelet activation with minimal requirement of ADP and TxA2. In contrast, ADP and TxA2 signaling are critical for formation of the outer shell region.15-17
Representative images of hemostatic plugs formed in WT and GRK2−/− mice at 1.5 minutes after injury are shown in Figure 2Ai. Platelet accumulation occurred at the same initial rate in GRK2−/− mice as in WT control mice. However, after reaching 40 seconds, there was a decline in CD41 fluorescence area in control mice that did not occur in the GRK2−/− mice (Figure 2Aii). Total platelet accumulation in the GRK2−/− mice was approximately one-third greater than in the control mice, as measured by area under the curve (Figure 2Aiii; supplemental Figure 3A). This increase was due to an expansion of the P-selectin (–) shell region, rather than the P-selectin (+) core region (Figure 2Aiv; supplemental Figure 3B). Fibrin accumulation was unaffected in GRK2−/− mice (Figure 2B; supplemental Figure 3C).
![Increased platelet accumulation at the site of vascular injury in GRK2−/− mice. (Ai) Representative images of hemostatic plugs formed in WT and GRK2−/− mice 1.5 minutes after injury. (Aii) Confocal intravital fluorescence microscopy was performed to follow platelet accumulation (CD41) and P-selectin expression over 3 minutes. (Aiii) Platelet accumulation at the site of injury as measured by CD41 (area under the curve [AUC]) for 61 injuries in 7 WT mice and 41 injuries in 7 GRK2−/− mice. (Aiv) P-selectin positivity expressed as AUC. Forty-three injuries in 7 WT mice and 35 injuries in 7 GRK2−/− mice. (Bi) Fibrin deposition was examined after making small penetrating injuries in the cremaster muscle arterioles with a laser in GRK2−/− mice and littermate control mice. Thirty injuries in 7 WT mice and 29 injuries in 7 GRK2−/− mice. (Bii) Fibrin accumulation as measured by the mean fibrin area at the end of the 3-minute interval. Data sets were compared by using a two-by-two unbalanced analysis of variance. Data are presented as mean ± standard error of the mean.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/15/10.1182_bloodadvances.2022007007/3/m_advancesadv2022007007f2.png?Expires=1765936306&Signature=L2s9ZI8aVInCs4SAY8TAIw9ZW6s4nvf4yQQ72Up6lKkVmdCs6rh1~LxZEZFUTZGnr5dRBDHQAzJ3TkaKZzLf-HwwUGiuGnS9Eg9DnH-ZoaiQ1sG27EHjPkQjQ7Xf8SVrYxCJxHJU5Znvh~3oajraNNSNWSead0CFgxDE3k-sgLtVDLkZPq4CZQstHoFVlIzoDNPSH1HCvLT~qAviEtV6ceeTB2cZskofz5ZETWlCrtej-DgAdo0uCnJq-6oX0ljCPpSFGgh7c9tyWp8QCzR8bfbzw1PNljVHD4geqvwwB72uLXNnq6OPFLWRSZ1i56n0IfwgI8bT6pAkoSSGKnr75A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Increased platelet accumulation at the site of vascular injury in GRK2−/− mice. (Ai) Representative images of hemostatic plugs formed in WT and GRK2−/− mice 1.5 minutes after injury. (Aii) Confocal intravital fluorescence microscopy was performed to follow platelet accumulation (CD41) and P-selectin expression over 3 minutes. (Aiii) Platelet accumulation at the site of injury as measured by CD41 (area under the curve [AUC]) for 61 injuries in 7 WT mice and 41 injuries in 7 GRK2−/− mice. (Aiv) P-selectin positivity expressed as AUC. Forty-three injuries in 7 WT mice and 35 injuries in 7 GRK2−/− mice. (Bi) Fibrin deposition was examined after making small penetrating injuries in the cremaster muscle arterioles with a laser in GRK2−/− mice and littermate control mice. Thirty injuries in 7 WT mice and 29 injuries in 7 GRK2−/− mice. (Bii) Fibrin accumulation as measured by the mean fibrin area at the end of the 3-minute interval. Data sets were compared by using a two-by-two unbalanced analysis of variance. Data are presented as mean ± standard error of the mean.
Increased platelet accumulation at the site of vascular injury in GRK2−/− mice. (Ai) Representative images of hemostatic plugs formed in WT and GRK2−/− mice 1.5 minutes after injury. (Aii) Confocal intravital fluorescence microscopy was performed to follow platelet accumulation (CD41) and P-selectin expression over 3 minutes. (Aiii) Platelet accumulation at the site of injury as measured by CD41 (area under the curve [AUC]) for 61 injuries in 7 WT mice and 41 injuries in 7 GRK2−/− mice. (Aiv) P-selectin positivity expressed as AUC. Forty-three injuries in 7 WT mice and 35 injuries in 7 GRK2−/− mice. (Bi) Fibrin deposition was examined after making small penetrating injuries in the cremaster muscle arterioles with a laser in GRK2−/− mice and littermate control mice. Thirty injuries in 7 WT mice and 29 injuries in 7 GRK2−/− mice. (Bii) Fibrin accumulation as measured by the mean fibrin area at the end of the 3-minute interval. Data sets were compared by using a two-by-two unbalanced analysis of variance. Data are presented as mean ± standard error of the mean.
Deletion of GRK2 in mouse platelets increases the response to ADP stimulation ex vivo
Next, we compared platelet integrin activation and α-granule secretion (P-selectin exposure) between GRK2−/− and WT mice to further examine the effect of GRK2 deletion on platelet activity. Using flow cytometry to measure the activated integrin αIIbβ3 and P-selectin in highly diluted platelets with their specific antibodies (Jon/A and anti–P-selectin, respectively), we observed significantly increased integrin αIIbβ3 activation and P-selectin exposure in GRK2−/− platelets in response to ADP stimulation. There was a slightly increased P-selectin exposure in GRK2−/− platelets in response to U46619 (TxA2 analogue) (Figure 3A; supplemental Figure 4). Platelet activation in response to PAR4 thrombin receptor agonist peptide (PAR4-AP), AYPGKF, and convulxin (glycoprotein VI ligand) was normal (Figure 3A). In addition, no increase in basal P-selectin exposure or integrin activation on GRK2−/− platelets was noted, suggesting that the GRK2−/− platelets are not circulating in a preactivated state.
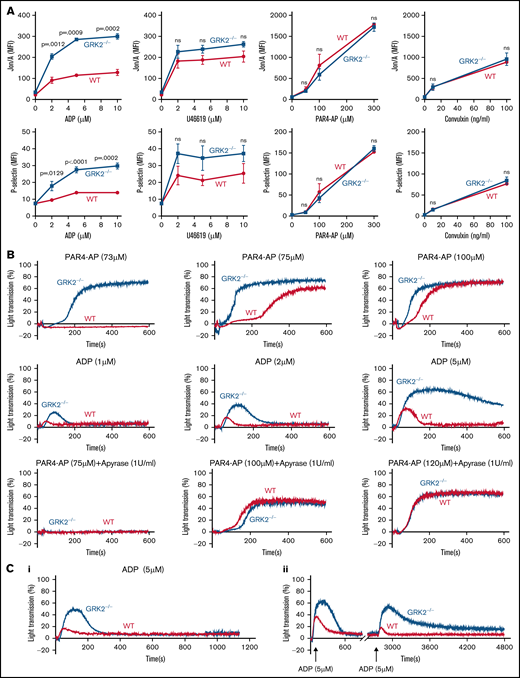
Increased platelet integrin activation, α-granule secretion, aggregation, and decreased receptor desensitization in GRK2−/− mice under ADP stimulation. (A) Platelets from GRK2−/− and littermate control (WT) mice were stained with fluorophore-conjugated antibodies to either activated integrin αIIbβ3 (Jon/A antibody) or P-selectin and measured by flow cytometry. Platelets were stimulated with ADP, TxA2 mimetic (U46619), PAR4-AP (AYPGKF), or convulxin at the concentrations indicated (n = 4). (B) Platelet aggregation in response to a PAR4-AP or ADP at the concentrations indicated (n ≥ 3). (C) Receptor desensitization measured by platelet aggregation in response to ADP. (Ci) Platelets from GRK2−/− and littermate WT control mice were stimulated with 5 µM ADP after pre-incubation with 5 µM ADP. (Cii) Platelets from GRK2−/− and littermate WT control mice were stimulated with 5 µM ADP first and restimulated with 5 µM ADP after a 40-minute interval. n = 4. P ≤ .05 was considered statistically significant. MFI, mean fluorescence intensity; ns, not significant.
Increased platelet integrin activation, α-granule secretion, aggregation, and decreased receptor desensitization in GRK2−/− mice under ADP stimulation. (A) Platelets from GRK2−/− and littermate control (WT) mice were stained with fluorophore-conjugated antibodies to either activated integrin αIIbβ3 (Jon/A antibody) or P-selectin and measured by flow cytometry. Platelets were stimulated with ADP, TxA2 mimetic (U46619), PAR4-AP (AYPGKF), or convulxin at the concentrations indicated (n = 4). (B) Platelet aggregation in response to a PAR4-AP or ADP at the concentrations indicated (n ≥ 3). (C) Receptor desensitization measured by platelet aggregation in response to ADP. (Ci) Platelets from GRK2−/− and littermate WT control mice were stimulated with 5 µM ADP after pre-incubation with 5 µM ADP. (Cii) Platelets from GRK2−/− and littermate WT control mice were stimulated with 5 µM ADP first and restimulated with 5 µM ADP after a 40-minute interval. n = 4. P ≤ .05 was considered statistically significant. MFI, mean fluorescence intensity; ns, not significant.
A platelet aggregation assay further verified this result, showing that GRK2−/− mouse platelets had an increased response to ADP stimulation (Figure 3B). Because other agonists rely on feedback from ADP signaling for maximal effect, we next performed platelet aggregation in response to PAR4-AP stimulation in the absence or presence of apyrase. There was an increase in platelet aggregation in response to PAR4-AP stimulation alone (Figure 3B; supplemental Figure 5A). However, the increased aggregation in GRK2−/− mouse platelets was abolished when secreted ADP was removed by treatment with 1 U/mL apyrase (Figure 3B; supplemental Figure 5B). Furthermore, the response to PAR4-AP stimulation was shifted to higher concentrations in the presence of apyrase, which is consistent with previous reports.18-20 Conversely, GRK2−/− platelet aggregation was normal in response to U46619 or convulxin stimulation (supplemental Figure 5C-D). We also used single end point luminescence assays6 to measure ATP release in platelets. There was no difference in ATP release between WT and GRK2−/− platelets in response to ADP, indicating that deletion of GRK2 in platelets did not affect dense granule secretion after ADP stimulation (supplemental Figure 6). Together, these observations suggest the specific targeting of GRK2 to the ADP-dependent signaling pathway.
GRK2 affects ADP signaling by receptor desensitization
GRK2 is known to modulate GPCR signaling pathways at the receptor level via phosphorylation-dependent β-arrestin recruitment and receptor endocytosis.8 To determine if GRK2 regulates ADP signaling through receptor desensitization, we measured platelet aggregation in response to ADP restimulation in WT and GRK2−/− mouse platelets under incubation or stirring conditions. After pre-incubation with 5 μM ADP for 3 minutes, stimulation with 5 μM ADP induced a greater aggregation response in GRK2−/− platelets than in WT platelets (Figure 3Ci). The second stimulation with 5 μM ADP after prechallenge also induced a greater aggregation response in GRK2−/− mouse platelets than in WT platelets (Figure 3Cii). ADP rechallenge caused a significant decrease in the maximum aggregation in WT platelets, whereas there was only a small decrease in GRK2−/− platelets after ADP restimulation (supplemental Figure 7). Together, these data suggest that GRK2 may regulate ADP receptors through the canonical mechanism of desensitization.
GRK2 deletion in mouse platelets affects P2Y1 receptor signaling pathways ex vivo
Human and mouse platelets express 2 distinct ADP receptors, P2Y1 and P2Y12. Coactivation of these 2 receptors is essential for ADP-induced platelet activation.21 P2Y1 couples to Gq and P2Y12 couples to Gi family members, primarily Gi2. P2Y1 activation leads to the release of Ca2+ from intracellular stores followed by the influx of extracellular Ca2+.22,23 P2Y1-dependent signaling mediates platelet shape change and transient, rapidly reversible aggregation induced by ADP. P2Y12 is critically involved in integrin activation, inhibition of cyclic adenosine monophosphate (cAMP) formation, and stabilization of platelet aggregation.24
To assess the role of GRK2 in P2Y1-mediated platelet activation, we first studied activation of WT platelets vs GRK2−/− platelets in response to MRS2365, an agonist specific to P2Y1, by flow cytometry.25,26 There was an increase in integrin activation and an increasing trend in P-selectin exposure in GRK2−/− platelets compared with WT platelets after stimulation with MRS2365 (Figure 4A; supplemental Figure 8). We next measured intracellular Ca2+ release in WT and GRK2−/− mouse platelets in response to ADP or MRS2365. It is worth noting that Ca2+ mobilization in response to ADP is a readout for P2Y1- and not for P2Y12-mediated signaling.3 There was a greater increase in intracellular Ca2+ concentration in the GRK2−/− platelets relative to WT platelets, after stimulation with ADP or MRS2365 (Figure 4B). Taken together, these results suggest that GRK2 negatively regulates P2Y1-mediated signaling during platelet activation.
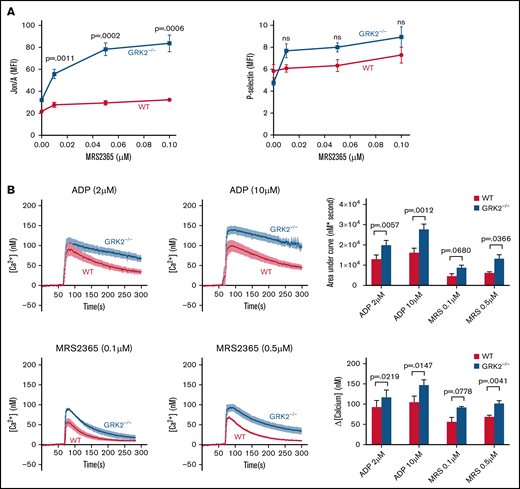
Increased platelet activation and calcium mobilization downstream of P2Y1-dependent signaling in GRK2−/− platelets. (A) Platelet activation. Platelets from GRK2−/− and littermate control mice (WT) were stained with fluorophore-conjugated antibodies to either activated integrin αIIbβ3 (Jon/A antibody) or P-selectin and measured by flow cytometry. Platelets were stimulated with MRS2365 (MRS). n = 4. (B) Calcium mobilization. Mouse platelets were loaded with fura-2 AM (10 μM) and stimulated with ADP or MRS at the concentrations indicated in the absence of extracellular Ca2+. Representative measurements are shown. The results of 6 experiments, ADP (upper), and the results of 4 experiments, MRS (lower), are summarized (mean ± standard error of the mean). MFI, mean fluorescence intensity; ns, not significant.
Increased platelet activation and calcium mobilization downstream of P2Y1-dependent signaling in GRK2−/− platelets. (A) Platelet activation. Platelets from GRK2−/− and littermate control mice (WT) were stained with fluorophore-conjugated antibodies to either activated integrin αIIbβ3 (Jon/A antibody) or P-selectin and measured by flow cytometry. Platelets were stimulated with MRS2365 (MRS). n = 4. (B) Calcium mobilization. Mouse platelets were loaded with fura-2 AM (10 μM) and stimulated with ADP or MRS at the concentrations indicated in the absence of extracellular Ca2+. Representative measurements are shown. The results of 6 experiments, ADP (upper), and the results of 4 experiments, MRS (lower), are summarized (mean ± standard error of the mean). MFI, mean fluorescence intensity; ns, not significant.
GRK2 deletion in mouse platelets affects P2Y12 receptor signaling pathways ex vivo
To determine the role of GRK2 in P2Y12 receptor signaling, we investigated several P2Y12-dependent signaling pathways in WT and GRK2−/− platelets. First, we studied the activation of the small GTPase RAS-related protein 1 (RAP1) in WT and GRK2−/− platelets. RAP1 activation is one of the critical steps leading to integrin αIIbβ3 activation in platelets.27,28 P2Y12-mediated signaling plays a critical role in maintaining sustained RAP1 activation.29 Our data showed no increase in basal RAP1-GTP level in GRK2−/− platelets compared with that of WT mice. In contrast, there was a significantly greater increase in RAP1-GTP levels in response to ADP stimulation in GRK2−/− platelets than in controls (Figure 5A).
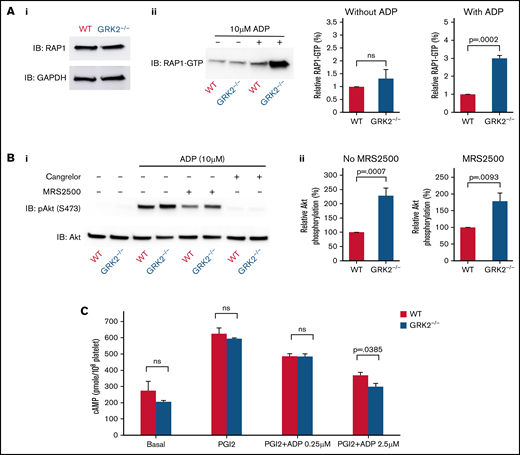
Increased RAP1 activation, Akt phosphorylation, and reduced cAMP formation downstream of P2Y12-dependent signaling in GRK2−/− platelets. (A) RAP1 activation. Ai: RAP1 protein expression in WT and GRK2−/− platelets. Aii: RAP1-GTP levels were measured in platelets that were either unstimulated or stimulated with ADP for 5 minutes (mean ± standard error of the mean; n = 4). (B) Akt phosphorylation. Gel-filtered platelets from GRK2−/− mice or matched WT controls were lysed directly or incubated with 10 μM ADP for 5 minutes in the presence or absence of the P2Y1 antagonist MRS2500 (50 mM) or the P2Y12 antagonist cangrelor (100 nM), as indicated. (Bi) Lysates were probed with anti–p-Akt (S473) and reprobed with anti-Akt antibody. (Bii) The p-Akt signal was normalized to the Akt loading control and is represented as signal relative to that of the WT with or without antagonist MRS2500 (mean ± standard error of the mean; n = 6). (C) cAMP formation. cAMP levels in resting platelets and platelets stimulated with PGI2 in the presence or absence of ADP at the final concentrations indicated (mean ± standard error of the mean; n = 3). ns, not significant.
Increased RAP1 activation, Akt phosphorylation, and reduced cAMP formation downstream of P2Y12-dependent signaling in GRK2−/− platelets. (A) RAP1 activation. Ai: RAP1 protein expression in WT and GRK2−/− platelets. Aii: RAP1-GTP levels were measured in platelets that were either unstimulated or stimulated with ADP for 5 minutes (mean ± standard error of the mean; n = 4). (B) Akt phosphorylation. Gel-filtered platelets from GRK2−/− mice or matched WT controls were lysed directly or incubated with 10 μM ADP for 5 minutes in the presence or absence of the P2Y1 antagonist MRS2500 (50 mM) or the P2Y12 antagonist cangrelor (100 nM), as indicated. (Bi) Lysates were probed with anti–p-Akt (S473) and reprobed with anti-Akt antibody. (Bii) The p-Akt signal was normalized to the Akt loading control and is represented as signal relative to that of the WT with or without antagonist MRS2500 (mean ± standard error of the mean; n = 6). (C) cAMP formation. cAMP levels in resting platelets and platelets stimulated with PGI2 in the presence or absence of ADP at the final concentrations indicated (mean ± standard error of the mean; n = 3). ns, not significant.
P2Y12 signaling in platelets triggers Gi-mediated activation of the serine-threonine kinases Akt1 and Akt2.30,31 Thus, we compared phosphorylation of Akt (p-Akt) between ADP-stimulated WT and GRK2−/− mouse platelets in the presence or absence of MRS2500 (P2Y1 antagonist) or cangrelor (P2Y12 antagonist). GRK2−/− platelets showed a significant increase in p-Akt upon ADP stimulation (Figure 5B), and this difference persisted when blocking P2Y1 receptor with MRS2500. However, the p-Akt level in both WT and GRK2−/− platelets was abolished when platelets were incubated with the P2Y12 receptor antagonist cangrelor. Interestingly, in the presence of MRS2500, there was a small reduction in p-Akt after ADP stimulation, which may be due to indirect contributions from P2Y12-independent pathways.
Endothelium-derived prostaglandin (PGI2) targets the prostacyclin receptor, resulting in an increase in cAMP to prevent premature platelet activation. During platelet activation, cAMP production is suppressed by ADP-triggered P2Y12 signaling via Gi2α. To examine whether GRK2 deletion affects P2Y12 inhibition of cAMP accumulation, we measured the cAMP level in both WT and GRK2−/− platelets under resting and PGI2 conditions with or without ADP stimulation. These data show that there was no difference in basal and PGI2-induced cAMP formation in WT controls vs GRK2−/− platelets in the absence of ADP stimulation. In contrast, a slight but significant reduction in cAMP formation was observed in GRK2−/− platelets compared with the controls after ADP stimulation (Figure 5C). Collectively, these results show that GRK2 negatively regulates P2Y12-mediated signaling pathway in platelets.
GRK2 regulates P2Y12 signaling during hemostatic plug formation in vivo
Our prior studies show the importance of ADP/P2Y12 signaling for platelet recruitment and/or retention in the outer shell region of hemostatic plugs.15,16 We therefore hypothesize that blocking P2Y12 signaling would eliminate the observed increased platelet accumulation at the outer layer of the thrombus in GRK2−/− mice. To test this hypothesis, we examined thrombus formation after laser-induced injury to the cremaster muscle arterioles in WT and GRK2−/− mice treated with a P2Y12 antagonist (cangrelor). Cangrelor or vehicle (saline) was administered IV immediately before each laser injury, as described in the Materials and methods. Platelet accumulation in saline-treated groups displayed a pattern similar to that shown in Figure 2Aii. Cangrelor treatment resulted in a reduction in total platelet accumulation at the site of vascular injury (Figure 6A; supplemental Figure 9). The rates of platelet accumulation in WT vs GRK2−/− in the presence of cangrelor were virtually identical. Treatment with cangrelor did not attenuate formation of the hemostatic plug core region of P-selectin–positive platelets in both WT and GRK2−/− mice (supplemental Figure 10A). These results indicate that blocking P2Y12 receptors with cangrelor eliminates the difference in platelet accumulation between GRK2−/− mice and control mice at the site of vascular injury.
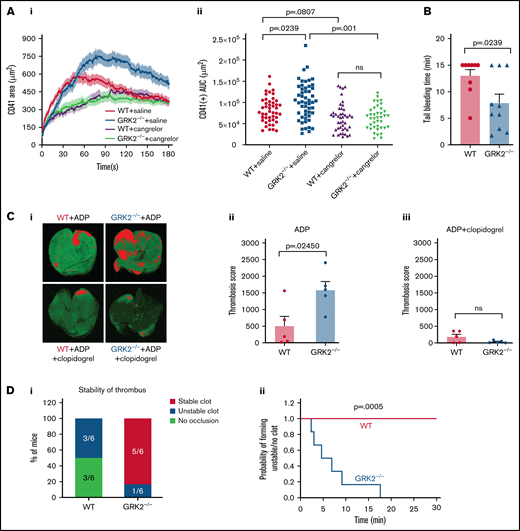
Deletion of GRK2 in platelets causes increased platelet accumulation in the shell region, shortens tail bleeding time, and enhances thrombosis. P2Y12 inhibition eliminates the phenotypic difference at the site of injury. (A) Laser-induced thrombus formation in WT and GRK2−/− mice treated with the P2Y12 antagonist (cangrelor). Cangrelor or vehicle (saline) was administered by IV immediately before each of the laser injuries. (Ai) Confocal intravital fluorescence microscopy was then performed to follow platelet accumulation (CD41) over 3 minutes. (Aii) Platelet accumulation at the site of injury as measured by CD41 of 46 injuries in 3 WT mice treated with saline, 52 injuries in 4 GRK2−/− mice treated with saline, 44 injuries in 4 WT mice treated with cangrelor, and 39 injuries in 4 GRK2−/− mice treated with cangrelor. Data sets were compared by using an unbalanced two-way analysis of variance with Tukey’s post hoc test. (B) Hemostatic response was measured by tail bleeding. Tails of anesthetized mice were transected and the time to complete arrest of bleeding (as defined by no bleeding recurring within 2 minutes) was recorded for each mouse. n = 9 mice per genotype. (Ci) Representative images of anti-glycoprotein IX (GPIX)–labeled thrombi in lungs harvested from WT and GRK2−/− mice treated with ADP in the absence or presence of clopidogrel (P2Y12 antagonist). (Cii and Ciii) Thrombosis scores, representing the mean thrombus area and number of thrombi, of 5 WT mice vs 5 GRK2−/− mice are summarized in each group. P ≤ .05 was considered statistically significant. (D) The carotid arteries of WT and GRK2−/− mice were injured by 7.5% FeCl3. Flow rates through the carotid artery were measured with a Doppler flow probe after vascular injury. (Di) Quantitation of the percentage of mice that formed a stable clot, unstable clot, or no occlusion. A χ2 test was conducted, producing a P value of .011. (Dii) The probability of forming unstable occlusion at a respective time interval for WT or GRK2−/− mice. Experiments were terminated at 30 minutes. Kaplan-Meier curves were analyzed by using the log-rank test. n = 6 mice per genotype. Data are presented as mean ± standard error of the mean. AUC, area under the curve. ns, not significant.
Deletion of GRK2 in platelets causes increased platelet accumulation in the shell region, shortens tail bleeding time, and enhances thrombosis. P2Y12 inhibition eliminates the phenotypic difference at the site of injury. (A) Laser-induced thrombus formation in WT and GRK2−/− mice treated with the P2Y12 antagonist (cangrelor). Cangrelor or vehicle (saline) was administered by IV immediately before each of the laser injuries. (Ai) Confocal intravital fluorescence microscopy was then performed to follow platelet accumulation (CD41) over 3 minutes. (Aii) Platelet accumulation at the site of injury as measured by CD41 of 46 injuries in 3 WT mice treated with saline, 52 injuries in 4 GRK2−/− mice treated with saline, 44 injuries in 4 WT mice treated with cangrelor, and 39 injuries in 4 GRK2−/− mice treated with cangrelor. Data sets were compared by using an unbalanced two-way analysis of variance with Tukey’s post hoc test. (B) Hemostatic response was measured by tail bleeding. Tails of anesthetized mice were transected and the time to complete arrest of bleeding (as defined by no bleeding recurring within 2 minutes) was recorded for each mouse. n = 9 mice per genotype. (Ci) Representative images of anti-glycoprotein IX (GPIX)–labeled thrombi in lungs harvested from WT and GRK2−/− mice treated with ADP in the absence or presence of clopidogrel (P2Y12 antagonist). (Cii and Ciii) Thrombosis scores, representing the mean thrombus area and number of thrombi, of 5 WT mice vs 5 GRK2−/− mice are summarized in each group. P ≤ .05 was considered statistically significant. (D) The carotid arteries of WT and GRK2−/− mice were injured by 7.5% FeCl3. Flow rates through the carotid artery were measured with a Doppler flow probe after vascular injury. (Di) Quantitation of the percentage of mice that formed a stable clot, unstable clot, or no occlusion. A χ2 test was conducted, producing a P value of .011. (Dii) The probability of forming unstable occlusion at a respective time interval for WT or GRK2−/− mice. Experiments were terminated at 30 minutes. Kaplan-Meier curves were analyzed by using the log-rank test. n = 6 mice per genotype. Data are presented as mean ± standard error of the mean. AUC, area under the curve. ns, not significant.
Absence of GRK2 accelerates pulmonary thromboembolism and promotes thrombosis in FeCl3-induced carotid artery
Next, we examined the effects of GRK2 deficiency on platelet function using a pulmonary thromboembolism mouse model, a thrombosis model in which platelet activation is the most prominent feature.33 To determine whether deletion of GRK2 might increase the incidence of formation of pulmonary microthrombi induced by platelet activation, GRK2−/− and their WT control littermates were injected IV with 0.3 mg/g ADP. ADP has been previously shown to induce disseminated pulmonary thromboembolism.34,35 The result indicates that GRK2−/− mice had more platelet accumulation in the lungs than observed in the WT control mice (Figure 6Ci-ii). There was no difference in mortality rate between WT and GRK2−/− mice (supplemental Figure 10B). Blocking P2Y12 receptors with clopidogrel abrogated ADP-induced platelet activation and abolished the difference in platelet accumulation in the lungs (Figure 6C; supplemental Figure 10C). These results suggest that GRK2 plays an important role in limiting platelet accumulation in the lung.
To further determine the role of GRK2 in thrombosis, we performed a FeCl3-induced carotid artery injury model.36 Five of 6 GRK2−/− mice reached stable occlusion after 7.5% FeCl3-induced vascular injury. The WT control mice either did not occlude at all or had an unstable occlusion during the 30 minutes of the experiment (Figure 6D). These results indicate that GRK2 plays an important role in regulating thrombosis.
GRK2 does not bind with the PAR1 receptor in thrombin-activated human platelets
To examine whether GRK2 regulates thrombin signaling through a canonical interaction with PAR1, we attempted to immunoprecipitate GRK2 with anti-PAR1 antibody from either resting or thrombin-stimulated human platelets. In contrast to the increased binding of GRK6 to PAR1 as we previously reported (supplemental Figure 11A),3 PAR1 receptor does not bind GRK2 in activated platelets (supplemental Figure 11B). This finding is consistent with the functional readouts from flow cytometry and platelet aggregation assays in which deletion of GRK2 in mouse platelets does not affect PAR-dependent signaling.
Endogenous GRK2 in human platelets interacts with Gβγ subunits but not Gqα
As shown previously in nucleated cells, the RGS domain of GRK2 can bind with Gqα subunits, but not Giα subunits, in vitro and therefore inhibit Gq-mediated signaling.37 We observed the interaction between GRK2 and Gqα in human and mouse platelets using an in vitro glutathione S-transferase–GRK2 pull-down assay (Figure 7Ai). There was no binding of GRK2 to Gi2α. We also tested whether GRK2 interacts with endogenous Gqα in platelets. GRK2 was precipitated from resting and thrombin-activated human platelets with an anti-GRK2 antibody, and samples were probed for Gqα. No binding of GRK2 to endogenous Gqα was detected by immunoprecipitation (supplemental Figure 12A), even in the presence AlF4– to mimic the GTP hydrolysis transition state of Gqα (Figure 7Aii), suggesting that Gqα is not a direct target of negative regulation by GRK2 in platelets. Furthermore, membrane association of GRK2 is mediated by binding to Gβγ via its pleckstrin homology domain in nucleated cells, and this binding is critical for the activation of GRK2.38,39 We observed that GRK2 binds to subunits of Gβγ during platelet activation in response to thrombin stimulation but not to 2MesADP (Figure 7B; supplemental Figure 12B).
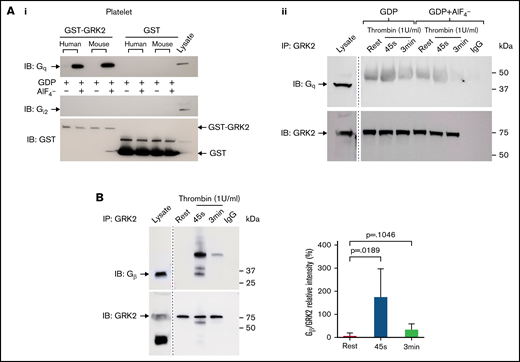
Gβ interacts with endogenous GRK2 in platelets ex vivo but Gqα does not. (Ai) GST-GRK2 was immunoprecipitated from human or mouse platelets, and samples were probed for Gqα or Giα2. Glutathione S-transferase (GST) reprobe indicated the equal loading. n = 2. (Aii) Human platelet lysate from resting or 1 U/mL thrombin-stimulated platelets in the presence or absence of GDP+AlF4– were subjected to immunoprecipitation of GRK2 and probed for Gq. n = 3. (B) GRK2 from resting and thrombin-stimulated (1 U/mL) human platelet lysate was immunoprecipitated with the specific mouse anti-GRK2 antibody and probed for Gβ. GRK2 was reprobed with an anti-GRK2 antibody. n = 6. A marker line was excised as indicated by the vertical line.
Gβ interacts with endogenous GRK2 in platelets ex vivo but Gqα does not. (Ai) GST-GRK2 was immunoprecipitated from human or mouse platelets, and samples were probed for Gqα or Giα2. Glutathione S-transferase (GST) reprobe indicated the equal loading. n = 2. (Aii) Human platelet lysate from resting or 1 U/mL thrombin-stimulated platelets in the presence or absence of GDP+AlF4– were subjected to immunoprecipitation of GRK2 and probed for Gq. n = 3. (B) GRK2 from resting and thrombin-stimulated (1 U/mL) human platelet lysate was immunoprecipitated with the specific mouse anti-GRK2 antibody and probed for Gβ. GRK2 was reprobed with an anti-GRK2 antibody. n = 6. A marker line was excised as indicated by the vertical line.
Pharmacologic inhibition of GRK2 in human platelets
We next investigated the effect of inhibiting GRK2 activity on platelet activation using a GRK2-specific inhibitor, Compound 101 (Cmpd101). Cmpd101 is a potent and selective GRK2 inhibitor and has been widely used in studying the roles of GRK2 in GPCR desensitization.40 The 50% inhibitory concentration value of Cmpd101 to inhibit GRK2-mediated bovine rod outer segment (bROS) phosphorylation is 290 nM. Treatment of human platelets with Cmpd101 results in an increase in platelet activation in response to 2MesADP (supplemental Figure 13), suggesting that GRK2 activity is required in ADP-mediated signaling in human platelets.
Discussion
Dysregulated platelet activation and thrombus instability can lead to venous thrombosis, myocardial infarction, and stroke.41 Considerable efforts have been made to treat thrombotic disorders by selectively inhibiting the pathways downstream of the ADP-P2Y12 signaling pathway.42-44 To avoid the risk of premature platelet activation or excess platelet accumulation at sites of injury, several regulatory events are in place to limit the rate and/or extent of platelet activation, including regulators that can limit P2Y12-mediated platelet activation.29 Although GRK2 is a critical regulator of GPCR signaling in the heart, its contribution to platelet activation and its role in GPCR-dependent signaling in platelets are still poorly understood.
In this study, we asked if GRK2 functions as a checkpoint to limit the intensity and duration of platelet activation downstream of GPCRs. Taking advantage of a GRK2−/− mouse line in which GRK2 is specifically deleted in megakaryocyte/platelets, we have identified a unique regulatory role of GRK2 in platelets downstream of ADP signaling. Among all the agonists studied in platelet functional assays, only ADP stimulation causes an increase in GRK2−/− platelet activation over WT, suggesting a dispensable role of GRK2 in thrombin/PAR4- or TxA2/TP-mediated signaling. Human platelets express 150 copies of P2Y1 receptor and 400 copies of P2Y12 receptor per cell, respectively.45 In comparison, mouse platelets express 1000 copies of P2Y1 receptor and 2700 copies of P2Y12 receptor per cell.46 Both human and mouse platelets express higher levels of P2Y12 than that of P2Y1. Our studies show that deletion of GRK2 in mouse platelets leads to an increase in ADP-induced platelet activation, suggesting a critical role for GRK2 in ADP P2Y12-dependent signaling. These observations are consistent with the previous findings by Hardy et al11 that desensitization of P2Y12 is mediated by GRK2. In addition, they show that desensitization of P2Y1 receptor in 1321N1 human astrocytoma cells is mainly regulated by protein kinase C but not by GRK2. In contrast to their findings in 1321N1 cells, our studies show that knocking out GRK2 in platelets causes an increase in P2Y1-dependent platelet activation. These findings indicate that GRK2 negatively regulates P2Y1-mediated signaling in platelets. It is worth noting that 1321N1 cells express similar levels of P2Y1 and P2Y12 receptors,11 indicating that the regulation of P2Y1 by protein kinase C in nucleated cells is presumably not operative in anucleate platelets. Our results are consistent with a recent report showing that platelets expressing a mutant P2Y1 receptor that does not desensitize have increased platelet activation in response to ADP and MRS2365 in vitro. Interestingly, impaired P2Y1 receptor desensitization had no effect on in vivo hemostasis or thrombosis (Paul et al., ISTH 2021).47 In addition, recent studies using platelets deficient in arrestin-3 indicate that arrestin-3 negatively regulates platelet activation downstream of P2Y receptors, which is consistent with what we have observed about the regulatory roles of GRK2 in ADP/P2Y signaling.48,49
To further test whether the loss of GRK2 produces a pathway-selective gain of function in vivo, we investigated the consequence of GRK2 deficiency on thrombus formation using several injury models. In the laser-induced cremaster arteriole injury model, thrombin is the primary driver for platelet activation in the core of hemostatic thrombi, whereas ADP and TxA2 are the main drivers for platelet accumulation in the shell.15 Hemostatic plugs formed in GRK2−/− mice have a larger shell region, with no increase in the size of the core region. In addition, GRK2−/− mice have more platelet accumulation in the lungs after ADP stimulation than observed in the WT control mice. Pharmacologic inhibition of P2Y12 signaling with cangrelor or clopidogrel abrogates the phenotypic difference between WT control and GRK2−/− mice. These findings suggest that the enhanced platelet accumulation observed in GRK2−/− mice is primarily due to loss of GRK2-mediated regulation of P2Y12 signaling. However, we are unable to examine direct interaction between GRK2 and P2Y12 due to the lack of an effective P2Y12 antibody, either for western blot analysis or for immunoprecipitation.
The multifunctionality of GRK2 has recently gained attention due to a constellation of effects, including both its canonical (GRK2-mediated phosphorylation) and noncanonical (kinase-independent molecular interactions) functions.50,51 For example, it has been shown in nucleated cells that GRK2 is recruited to the membrane and binds activated Gqα via its RGS domain.37,52 However, the lack of endogenous binding of Gqα by GRK2 in platelets is consistent with a previous report regarding the lack of binding of Gqα by GRK2 in the heart.37 In addition, GRK2 contains a C-terminal pleckstrin homology domain, which allows for Gβγ binding after dissociation of the heterotrimeric G protein. The binding of GRK2 with Gβγ facilitates membrane targeting of GRK2, which also stimulates phosphorylation of activated GPCRs.53 We were able to observe this interaction in activated platelets, and this interaction may have consequences for the activation of GRK2. In cells other than platelets, GRK2 has been reported to interact directly with phosphatidylinositol 3-kinase γ54,55 and Akt.56 These binding partners of GRK2 may indicate the noncanonical functions of GRK2 in regulating GPCR signaling, which might contribute to the observed distinct functions of GRK2 from other GRKs, such as GRK5 and GRK6. Elucidation of novel GRK2 substrates, other GRK2-mediated signaling events, and GRK2’s noncanonical functions and binding partners in platelets is worthy of investigation in a future study.
In summary, we have shown for the first time that GRK2 regulates the hemostatic response by limiting platelet activation via ADP P2Y1- and P2Y12-dependent signaling. The unique regulatory role of GRK2 in ADP signaling in platelets is highly relevant to recent efforts in developing small molecule GRK2 inhibitors in the treatment of heart disease. Our finding regarding the role of GRK2 in platelets highlights the potential for significant off-target effects of GRK2 inhibition, such as platelet hyperactivity and subsequent thrombosis, due to the loss of GRK2-mediated rate-limiting effects on ADP signaling in platelets. Thus, it emphasizes the benefits of maintaining a certain level of GRK2 activity in platelets in preventing thrombotic events. The efforts and knowledge gained in this study should enable better development of GRK2-based therapeutic options for the treatment of cardiovascular diseases and thrombotic disorders.
Acknowledgments
The authors thank Xi Chen for technical assistance.
These studies were supported by the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL144574, P.M.).
Authorship
Contributions: X.Z., M.C., J.V.M., Y.Y., and A.B. performed experiments; X.Z., M.C., J.K.C., J.V.M., W.J.K., S.E.M., M.T., T.J.S., L.Z., and P.M. designed experiments; and X.Z., and P.M. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Peisong Ma, Cardeza Foundation for Hematologic Research, Department of Medicine, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, PA 20036; e-mail: Peisong.Ma@jefferson.edu; and Li Zhu, Cyrus Tang Hematology Center, Soochow University, Suzhou, China; e-mail: zhul@suda.edu.cn.

