

Key Points
Stage I FL is a heterogeneous disease that has clear genomic and microenvironmental similarities with stage III/IV disease.
Stage I FL can be classified into 3 clusters, 2 of which display different underlying oncogenic pathways compared with stage III/IV FL.

Abstract
Although the genomic and immune microenvironmental landscape of follicular lymphoma (FL) has been extensively investigated, little is known about the potential biological differences between stage I and stage III/IV disease. Using next-generation sequencing and immunohistochemistry, 82 FL nodal stage I cases were analyzed and compared with 139 FL stage III/IV nodal cases. Many similarities in mutations, chromosomal copy number aberrations, and microenvironmental cell populations were detected. However, there were also significant differences in microenvironmental and genomic features. CD8+ T cells (P = .02) and STAT6 mutations (false discovery rate [FDR] <0.001) were more frequent in stage I FL. In contrast, programmed cell death protein 1–positive T cells, CD68+/CD163+ macrophages (P < .001), BCL2 translocation (BCL2trl+) (P < .0001), and KMT2D (FDR = 0.003) and CREBBP (FDR = 0.04) mutations were found more frequently in stage III/IV FL. Using clustering, we identified 3 clusters within stage I, and 2 clusters within stage III/IV. The BLC2trl+ stage I cluster was comparable to the BCL2trl+ cluster in stage III/IV. The two BCL2trl– stage I clusters were unique for stage I. One was enriched for CREBBP (95%) and STAT6 (64%) mutations, without BLC6 translocation (BCL6trl), whereas the BCL2trl– stage III/IV cluster contained BCL6trl (64%) with fewer CREBBP (45%) and STAT6 (9%) mutations. The other BCL2trl– stage I cluster was relatively heterogeneous with more copy number aberrations and linker histone mutations. This exploratory study shows that stage I FL is genetically heterogeneous with different underlying oncogenic pathways. Stage I FL BCL2trl– is likely STAT6 driven, whereas BCL2trl– stage III/IV appears to be more BCL6trl driven.
Introduction
Follicular lymphoma (FL) is the most common indolent non-Hodgkin lymphoma (NHL) in adults, with an incidence of 2.2 to 5 per 100 000 in the western world.1-3 The large majority of patients present with advanced-stage disease (stage III/IV) at diagnosis, whereas only 10% to 15% exhibit limited-stage disease at presentation.4,5
Patients with limited-stage FL, defined by stage I and limited, contiguous stage II disease, may be cured in 45% to 65% of cases with local radiotherapy (24 Gy involved-field radiotherapy) without further systemic treatment.5-11 Adding rituximab to chemotherapy has been shown to improve progression-free survival (PFS) but at the cost of mild toxicity and with conflicting results pertaining to improving overall survival (OS).11-13 Despite the responsiveness of advanced-stage FL to current chemo-immunotherapy modalities, the disease course is characterized by multiple relapses and is considered incurable.
The oncogenesis of FL suggests a primary systemic disease with BCL2 translocation (BCL2trl+) as an early transforming event, most likely occurring in the bone marrow and not in eventual presenting nodal sites. It is intriguing that a lymphoma characterized by a relapsing, protracted but eventually fatal course may be cured by local therapy only when presenting in the rare context of limited-stage disease. A key question therefore is whether limited-stage FL follows a different oncogenesis and is driven by specific genomic and/or microenvironmental features that may explain this distinctive clinical course.
The most characteristic genomic feature of FL is BCL2trl+, observed in 85% to 95% of advanced-stage FL but in only 42% to 50% of limited-stage FL.14,15 In cases in which this translocation has been identified, it results from an aberrant immunoglobulin locus rearrangement that occurs most frequently at the pre–B cell stage in the bone marrow and serves as one of the initiating events in FL oncogenesis. Whether other genomic differences occur in limited-stage FL compared with advanced-stage FL is currently unknown.
The interaction between tumor and immune microenvironmental cells in FL results in distinctive features and is likely to influence the clinical course and outcome in this disease.16 The role of specific immune microenvironment populations, such as T cells and macrophages, has not been fully elucidated. Despite extensive studies in advanced-stage FL, conflicting conclusions remain regarding the impact on survival.17-22 Again, there is a dearth of knowledge regarding the immune microenvironment of limited-stage FL. Only one study has reported microenvironment characteristics in different stages of FL. Stages I to IIIA were combined and considered early disease in that study, which was characterized by a significantly higher number of programmed cell death protein 1–positive (PD1+) T cells and a lower number of forkhead box P3–positive (FOXP3+) T cells compared with advanced-stage (stage IIIB-IV) disease.23
In a concerted effort, the Lunenburg Lymphoma Biomarker Consortium (LLBC) has collected a relatively large series of rigorously defined and clinically well-annotated cases of stage I nodal FL from clinical trial cohorts and a population-based registry. The genomic and immune microenvironmental characteristics of stage I FL were mapped, and subgroups were determined. Subsequently, this information was interpreted in the context of a large cohort of patients with advanced-stage FL collated in parallel by the LLBC members and analyzed with the same techniques.
Methods
Patient selection
Within the LLBC collaboration, samples were collected from 8 different cohorts. Stage I cases were collected from the European Organization for Research and Treatment of Cancer (EORTC) study 20971,24,25 the German Low-Grade Lymphoma Study Group (GLSG) early-stage FL study,26,27 and the Haematological Malignancy Research Network (HMRN) population-based registry.28 Detailed inclusion criteria and treatment protocols are provided in supplemental Table 1.
Stage III/IV cases were collected from the Lymphoma Study Association FL2000 study29,30 and the GLSG2000 study,31 together with the population-based registries from the HMRN and Sweden, and the institutional registries from St. Bartholomew’s Hospital and Stanford University Medical Center (detailed inclusion criteria and treatment protocols are provided in supplemental Table 1).
Patients for the stage I cohort, selected from the 2 studies and the population-based registry, needed to fulfill the following criteria: (1) stage I as determined by standard staging procedures at time of inclusion in study or database; (2) nodal, nonbulky disease (<7 cm); and (3) histologically confirmed FL grade 1 to 3A. Inclusion criteria for the patients in the stage III/IV cohort were: (1) stage III/IV disease as determined by standard staging procedures; (2) ≥5 nodal areas, with or without bulky disease; and (3) histologically confirmed FL grade 1 to 3A.
All stage I and III/IV cases were staged with computed tomography imaging and bone marrow biopsy. For both cohorts, availability of complete and detailed clinical information on demographic parameters, staging procedures, and treatment was required, as well as a representative diagnostic formalin-fixed paraffin-embedded biopsy sample.
Immunohistochemistry analysis of the microenvironment and tumor cells
Tissue microarrays were constructed centrally according to LLBC-validated protocols using duplicate cores of 1 mm in diameter.32 Sections of 3 µm were mounted on slides and stained for CD3, CD4, CD8, CD68, CD163, FOXP3, and PD1 according to standard procedures at the Barts Cancer Institute–Centre for Haemato-Oncology Research Laboratory (supplemental Table 2).
These microenvironment markers were scored on the whole core by a computerized system with automated scanning microscopy and computerized image analysis (Ariol SL-8, Leica Microsystems, Wetzlar, Germany) as validated in Sander et al32 and applied previously in Stevens et al.18 More detailed information is provided in the supplemental Methods.
In addition, tumor cell features were further assessed with immunohistochemistry (IHC) for expression of BCL2, with antibodies to different epitopes (DAKO124 and SP66), germinal center markers (BCL6, CD10, LMO2, and HGAL), a post–germinal center marker (MUM1), and a putative nodal marginal zone lymphoma marker (MNDA) (supplemental Table 2). MNDA was added as an extra control that these cases were true FL and not mantle zone lymphoma.
These IHC stains were performed according to standard procedures at the Department of Pathology, Amsterdam UMC location VUMC, Amsterdam, The Netherlands. All markers were independently scored on duplicate cores in a dichotomized manner as negative or positive, defined as >30% positive tumor cells, by 2 pathologists (D.d.J., B.S., or A.R.). All cores with <50% of scorable core surface area were excluded. In case of discordance between the 2 pathologists, a deciding score by the third pathologist was performed.
Gene mutation and copy number analysis using next-generation sequencing
Next-generation sequencing (NGS) library preparation and analysis were performed as previously described.33 Briefly, genomic DNA was extracted with the QIAamp DNA formalin-fixed paraffin-embedded Tissue Kit (Qiagen, Hilden, Germany) and fragmented by using a Covaris ME220 (Covaris Inc., Woburn, MA). Subsequently, NGS libraries were made with 100 ng sheared DNA and unique indexes (IDT, Coralville, IA) using the KAPA or KAPA Hyper Library Preparation kit (KAPA Biosystems, Wilmington, MA).
For copy number aberration (CNA), 50-bp single-read shallow whole-genome sequencing was performed on a HiSeq 4000 (Illumina, San Diego, CA). Sequence reads were aligned against the reference genome (GRCh37/hg19) with the Burrows-Wheeler Alignment tool (BWA aln; version 0.7.12)34 and de-duplicated with Picard tools (version 2.15). Copy number analysis was performed with QDNAseq (version 1.12.0),35 NoWaves (version 0.6),36 DNAcopy (version 1.50.1),37 ACE (version 0),38 CGHcall (version 2.38.0),39 and CGHregions (version 1.34).40
For mutation and translocation analysis, a 3Mb SeqCapEZ capture panel was designed in collaboration with Roche, containing coding regions of 369 genes and 12 genomic regions (Roche NimbleGen, Madison, WI; order ID 43712) (supplemental Tables 3 and 4); eight samples were equimolarly pooled to 1 µg for the capture, and 3 pools together were sequenced 150-bp paired-end on a HiSeq 4000 (Illumina).
Sequenced reads were trimmed with SeqPurge (version 0.1-104),41 aligned with BWA mem (version 0.7.12),34 realigned with ABRA (version 2.19),42 and duplicates removed with Picard tools (version 2.4.1; using the setting ASSUME SORT ORDER=queryname). Mutations were detected by LoFreq (version 2.1.3.1)43 and Mutect2 (version 4.1.7).44
Translocation detection was performed with BreaKmer (version 0.0.4), GRIDDS (version 1.4.2), Wham (version 1.7.0), and novoBreak (version 0.0.6).45-48 Translocations needed to be detected by at least 2 of the used tools. More detailed information is presented in the supplemental Methods.
Ethical committee statement
The study and protocols to obtain human archival tissues and patient data were approved by the local ethical committee of the VU University Medical Center, Amsterdam (FWA00017598), for all collaborating centers. They comply with the Code for Proper Secondary Use of Human Tissue in the Netherlands (http://www.fmwv.nl).
Statistical analysis
Clinical characteristics were summarized with descriptive statistics (median [range] for quantitative and frequency [percent] for qualitative variables) and compared by using χ2 or Mann-Whitney tests. Kaplan-Meier survival curves were constructed. PFS was defined as time from start of treatment to progression/transformation. OS was defined as time from start of treatment to death from any cause.
The average of the IHC biomarker scores from 2 cores was calculated. They were compared between groups of patients with a Kruskal-Wallis test corrected for multiple testing with the Benjamini-Hochberg method.
Comparison of frequencies of mutations and translocations was performed with Fisher’s exact test, and false discovery rates (FDRs) were controlled with the Benjamini-Hochberg method. P values and FDRs for comparisons between copy number regions were calculated with the R-package CGHtest.
Complete-linkage hierarchical clustering was performed with the function “hclust”’ of the “stat” package (https://stat.ethz.ch/R-manual/R-devel/library/stats/html/00Index.html). Features included in clustering were somatic mutations and focal and chromosomal arm–level aberrations present in >5% of the samples, and BCL2 and BCL6 translocations. The distance measure used for the clustering was defined as 1-corspearman for both the genes and the patient samples, implemented by the “cor” function, also from the “stat” package. The stability of the clusters was assessed by subsampling as described by Monti et al.49 All analyses were performed in R version 3.5.1 (R Foundation for Statistical Computing), and a two-sided P value <.05 was considered statistically significant.
Results
In total, 216 patients with stage I disease from 2 clinical trials and 1 population-based registry fulfilling the clinical selection criteria were included in this study. Complete targeted NGS data for mutations, translocations, and genome-wide copy number variations could be generated for 82 cases, 73 of which also had complete microenvironmental data (Figure 1A; supplemental Table 5).
![Outline of FL cases included in the study. A total of 233 stage I cases were initially submitted from 2 clinical trials and 1 population-based cohort; 216 fulfilled all clinical inclusion criteria (EORTC study 20971, n = 143; GLSG, rituximab, and involved-field radiotherapy in early-stage FL [MIR (Mabthera and Involved Field Radiation)] study, n = 39; and the HMRN population-based registry, n = 34). In 82 of 216 cases with targeted NGS data, a complete data set on translocation, CNA, and mutations was successfully obtained, meeting all quality measures (ie, sufficient amount of DNA [>100 ng] from formalin-fixed paraffin-embedded [FFPE] material, and sequencing results with a minimum mean target coverage off >30 reads for paired-end sequencing and 300 000 reads for shallow sequencing). For 73 of 82 cases, complete IHC data of 7 markers (CD3, CD4, CD8, PD1, FOXp3, CD68, and CD163) of the microenvironment were available, meeting all quality measures, indicating sufficient amount of FFPE material to obtain two 1-mm cores, and the cores should contain >50% tumor tissue to score. As a reference cohort, 667 stage III/IV cases were selected from 2 clinical trials and 4 population-based cohorts, of which 391 fulfilled all clinical inclusion criteria (LYSA [Lymphoma Study Association] FL2000 study, n = 163; GLSG2000 study, n = 98; HMRN population-based registry, n = 100; Sweden population-based registry, n = 19; St. Bartholomew’s Hospital, London, n = 6; and Stanford University Hospital, Stanford, n = 5). In 139 of 391 cases, complete NGS data meeting all quality measures were obtained. For 120 of 139 cases, complete microenvironment information meeting all quality measures was also available. Depicted in the green box are the cases that are incorporated in the analysis.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/18/10.1182_bloodadvances.2022008355/5/m_advancesadv2022008355f1.png?Expires=1767710556&Signature=Rfm54v8GulezBZ3Ts1MTJnH2R~~RYQFbHbe6azjAl4DpZEZMvow5vJqgAk8TGprGmBQUS-3tnoEAtuneVeCDietERDK~4Yjh139Hy0npmgdnBtk0PYkKmHM~i0rVv~8e4YSM2tdBqPll8eTLAerDAO4Hat9wR4VdfpZx29HQglvFVFVdMpqgXj9QIWOdUoRGKiGSwEcuC1IpTEOPmO1n2OjTOcwQchUIhYGLXne9Ju73eyy-ZsNkDzbUVLWqISlWhrpfSh0lMTcpUXkH1mCKfRzbzHoHFyFNKcVnedZysq42ej0Rg-5OMRk4XjonD4MMlssC-0w2kbCsAd~XszghdA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Outline of FL cases included in the study. A total of 233 stage I cases were initially submitted from 2 clinical trials and 1 population-based cohort; 216 fulfilled all clinical inclusion criteria (EORTC study 20971, n = 143; GLSG, rituximab, and involved-field radiotherapy in early-stage FL [MIR (Mabthera and Involved Field Radiation)] study, n = 39; and the HMRN population-based registry, n = 34). In 82 of 216 cases with targeted NGS data, a complete data set on translocation, CNA, and mutations was successfully obtained, meeting all quality measures (ie, sufficient amount of DNA [>100 ng] from formalin-fixed paraffin-embedded [FFPE] material, and sequencing results with a minimum mean target coverage off >30 reads for paired-end sequencing and 300 000 reads for shallow sequencing). For 73 of 82 cases, complete IHC data of 7 markers (CD3, CD4, CD8, PD1, FOXp3, CD68, and CD163) of the microenvironment were available, meeting all quality measures, indicating sufficient amount of FFPE material to obtain two 1-mm cores, and the cores should contain >50% tumor tissue to score. As a reference cohort, 667 stage III/IV cases were selected from 2 clinical trials and 4 population-based cohorts, of which 391 fulfilled all clinical inclusion criteria (LYSA [Lymphoma Study Association] FL2000 study, n = 163; GLSG2000 study, n = 98; HMRN population-based registry, n = 100; Sweden population-based registry, n = 19; St. Bartholomew’s Hospital, London, n = 6; and Stanford University Hospital, Stanford, n = 5). In 139 of 391 cases, complete NGS data meeting all quality measures were obtained. For 120 of 139 cases, complete microenvironment information meeting all quality measures was also available. Depicted in the green box are the cases that are incorporated in the analysis.
Outline of FL cases included in the study. A total of 233 stage I cases were initially submitted from 2 clinical trials and 1 population-based cohort; 216 fulfilled all clinical inclusion criteria (EORTC study 20971, n = 143; GLSG, rituximab, and involved-field radiotherapy in early-stage FL [MIR (Mabthera and Involved Field Radiation)] study, n = 39; and the HMRN population-based registry, n = 34). In 82 of 216 cases with targeted NGS data, a complete data set on translocation, CNA, and mutations was successfully obtained, meeting all quality measures (ie, sufficient amount of DNA [>100 ng] from formalin-fixed paraffin-embedded [FFPE] material, and sequencing results with a minimum mean target coverage off >30 reads for paired-end sequencing and 300 000 reads for shallow sequencing). For 73 of 82 cases, complete IHC data of 7 markers (CD3, CD4, CD8, PD1, FOXp3, CD68, and CD163) of the microenvironment were available, meeting all quality measures, indicating sufficient amount of FFPE material to obtain two 1-mm cores, and the cores should contain >50% tumor tissue to score. As a reference cohort, 667 stage III/IV cases were selected from 2 clinical trials and 4 population-based cohorts, of which 391 fulfilled all clinical inclusion criteria (LYSA [Lymphoma Study Association] FL2000 study, n = 163; GLSG2000 study, n = 98; HMRN population-based registry, n = 100; Sweden population-based registry, n = 19; St. Bartholomew’s Hospital, London, n = 6; and Stanford University Hospital, Stanford, n = 5). In 139 of 391 cases, complete NGS data meeting all quality measures were obtained. For 120 of 139 cases, complete microenvironment information meeting all quality measures was also available. Depicted in the green box are the cases that are incorporated in the analysis.
A cohort of 391 stage III/IV patients, who fulfilled the inclusion criteria, were selected from 2 clinical trials and 4 registries. For the final analysis, 139 of 391 cases with complete NGS data were included; 120 of these had complete microenvironment data available (Figure 1B; supplemental Table 5).
Clinical characteristics for the 82 stage I patients and 139 stage III/IV patients with complete NGS data are shown in Table 1. The study cohort is representative of the complete cohort of 602 patients with FL who fulfilled the initial clinical inclusion criteria (supplemental Table 6). Clinical variables such as presence of B-symptoms, higher Follicular Lymphoma International Prognostic Index score, low hemoglobin levels, and elevated lactate dehydrogenase levels were, as expected, significantly more frequent in the stage III/IV cohort. The 10-year PFS and OS of the stage I cohort were 56% and 83%, respectively (supplemental Figure 1).
Demographic and clinical characteristics of stage I and stage III/IV patients included for analysis in the study
| Characteristic | Stage I | Stage III/IV | P |
|---|---|---|---|
| (n = 82) | (n = 139) | ||
| Age at diagnosis | 58 | 57 | .96* |
| Median (range), Y | (28-85) | (27-95) | |
| Sex | |||
| Female | 32 (39%) | 62 (45%) | .42† |
| Male | 50 (61%) | 77 (55%) | |
| B-symptoms | <.001† | ||
| Present | 5 (6%) | 41 (30%) | |
| Absent | 77 (94%) | 97 (69%) | |
| Missing | – | 1 (1%) | |
| FLIPI risk categories | <.001† | ||
| Low | 68 (83%) | – | |
| Intermediate | 6 (7%) | 53 (38%) | |
| High | – | 77 (55%) | |
| Missing | 8 (10%) | 9 (7%) | |
| Hemoglobin | .002† | ||
| <12 g/L | 2 (2%) | 26 (19%) | |
| ≥12 g/L | 78 (95%) | 111 (80%) | |
| Missing | 2 (2%) | 2 (1%) | |
| Elevated lactate dehydrogenase | <.001† | ||
| Yes | 6 (7%) | 44 (32%) | |
| No | 71 (87%) | 94 (67%) | |
| Missing | 5 (6%) | 1 (1%) | |
| Stage | |||
| I | 82 (100%) | – | |
| III | – | 38 (27%) | |
| IV | – | 101 (73%) | |
| Bulky disease | |||
| <7 cm | 82 (100%) | 96 (69%) | |
| ≥7 cm | – | 38 (27%) | |
| Missing | – | 5 (4%) | |
| ECOG performance status | |||
| <2 | 81 (99%) | 128 (93%) | |
| ≥2 | – | 10 (7%) | |
| Missing | 1 (1%) | 1 (1%) | |
| Bone marrow involvement | |||
| Yes | – | 83 (60%) | |
| No | 82 (100%) | 49 (35%) | |
| Missing | – | 7 (5%) | |
| No. of involved nodal sites, median (range) | 1 (1-1) | 8 (5-14) | |
| First-line therapy | |||
| R-chemotherapy | 2 (2%) | 117 (84%) | |
| Chemotherapy | – | 5 (4%) | |
| IFRT | 38 (46%) | ||
| IFRT + TBI | 14 (17%) | ||
| IFRT + R | 23 (28%) | ||
| Watch and wait | – | 3 (2%) | |
| Other | 1 (1%)‡ | – | |
| Unknown | 4 (5%)§ | 14 (10%) | |
| Characteristic | Stage I | Stage III/IV | P |
|---|---|---|---|
| (n = 82) | (n = 139) | ||
| Age at diagnosis | 58 | 57 | .96* |
| Median (range), Y | (28-85) | (27-95) | |
| Sex | |||
| Female | 32 (39%) | 62 (45%) | .42† |
| Male | 50 (61%) | 77 (55%) | |
| B-symptoms | <.001† | ||
| Present | 5 (6%) | 41 (30%) | |
| Absent | 77 (94%) | 97 (69%) | |
| Missing | – | 1 (1%) | |
| FLIPI risk categories | <.001† | ||
| Low | 68 (83%) | – | |
| Intermediate | 6 (7%) | 53 (38%) | |
| High | – | 77 (55%) | |
| Missing | 8 (10%) | 9 (7%) | |
| Hemoglobin | .002† | ||
| <12 g/L | 2 (2%) | 26 (19%) | |
| ≥12 g/L | 78 (95%) | 111 (80%) | |
| Missing | 2 (2%) | 2 (1%) | |
| Elevated lactate dehydrogenase | <.001† | ||
| Yes | 6 (7%) | 44 (32%) | |
| No | 71 (87%) | 94 (67%) | |
| Missing | 5 (6%) | 1 (1%) | |
| Stage | |||
| I | 82 (100%) | – | |
| III | – | 38 (27%) | |
| IV | – | 101 (73%) | |
| Bulky disease | |||
| <7 cm | 82 (100%) | 96 (69%) | |
| ≥7 cm | – | 38 (27%) | |
| Missing | – | 5 (4%) | |
| ECOG performance status | |||
| <2 | 81 (99%) | 128 (93%) | |
| ≥2 | – | 10 (7%) | |
| Missing | 1 (1%) | 1 (1%) | |
| Bone marrow involvement | |||
| Yes | – | 83 (60%) | |
| No | 82 (100%) | 49 (35%) | |
| Missing | – | 7 (5%) | |
| No. of involved nodal sites, median (range) | 1 (1-1) | 8 (5-14) | |
| First-line therapy | |||
| R-chemotherapy | 2 (2%) | 117 (84%) | |
| Chemotherapy | – | 5 (4%) | |
| IFRT | 38 (46%) | ||
| IFRT + TBI | 14 (17%) | ||
| IFRT + R | 23 (28%) | ||
| Watch and wait | – | 3 (2%) | |
| Other | 1 (1%)‡ | – | |
| Unknown | 4 (5%)§ | 14 (10%) | |
ECOG, Eastern Cooperative Oncology Group; FLIPI, Follicular Lymphoma International Prognostic Index; IFRT, involved-field radiotherapy; PS, performance score; R, rituximab; TBI, total body irradiation.
P value per Mann-Whitney testing.
P value per χ2 testing.
Surgical removal.
IFRT without knowing if the patient received TBI or not.
The eight IHC markers (BCL2 DAKO124 and SPS66, CD10, BCL6, HGAL, LMO2, MNDA, and MUM1), used to confirm true stage I FL, could be evaluated in 75 of 82 patients. Concordance had to be reached with a third pathologist in 4% of the scored markers. In 91% of cases, a minimum of 3 of 4 germinal center markers were scored positive (CD10, 91%; BCL6, 96%; HGAL, 80%; and LMO2, 95%) (supplemental Table 7). Moreover, MNDA was not expressed in any of the cases,50 nor was MUM1, underlining the germinal center features. These findings exclude alternative diagnoses such as nodal marginal zone lymphoma and support classification of FL in all included cases.
Immune microenvironment in stage I and stage III/IV FL
Microenvironment analysis was available for 193 of 221 cases with complete NGS data (stage I, n = 73; stage III/IV, n = 120). In stage I disease, a significantly denser infiltrate of CD8+ cytotoxic T cells was observed (median stage I 13.7% vs stage III/IV 10.9%; P = .02), whereas PD1+ follicular T-helper cells (median stage I 1.8% vs stage III/IV 4.7%; P < .001) and macrophages (CD68+ median stage I 2.7% vs stage III/IV 3.6% [P < .001] and CD163+ median stage I 2.3% vs stage III/IV 4.1% [P < .001]) were more frequent in stage III/IV disease. Other cell populations as measured by T-cell markers CD3, CD4, and FOXP3 exhibited no significant differences (Figure 2A; supplemental Table 8). It should be noted, however, that although statistically significant differences were observed for CD8+and PD1+ T-cell populations and macrophage contribution, the absolute differences were minor and may only be appreciated by using automated image analysis.
![Microenvironment, mutations, translocations, and copy number landscape of stage I vs stage III/IV FL. (A) Percentage of positive nucleated cells for CD3, CD4, CD8, FOXP3, and PD1 depicted as boxplots. For CD163 and CD68, the percentage of positive area of the total cell area was computer-assisted scored are plotted in the boxplots (stage I in red [n = 73] and stage in blue III/IV [n = 120]). Significant differences are seen for CD8, PD1, CD163, and CD68 with a P value adjusted for multiple testing: *P = .02, **P = .002, ***P < .001. (B) Comparison plots for CNAs between stage I as filled areas (n = 82) and stage III/IV as lines (n = 139) are percentages of the number of cases with gains (positive value, blue) and losses (negative value, red), sorted for chromosome position (x-axis). (C) Frequency plots with P values (blue) calculated with a 2-sided rank-sum test with 10 000 permutations and FDR (striped segments) of the difference in CNAs; the horizontal dotted lines show the significance threshold P values <.05 in blue, and the FDR <0.1 in red. If the difference in CNA level crosses the P value, and the FDR level is <0.1, the difference is considered significant, which is indicated by * 6q23.3 and 9p21-22 (*P < .05). (D) Frequency of BLC2 and BLC6 translocations and top 20 mutated genes according to stage I in red (n = 82) and stage III/IV in blue (n = 139); significant differences are indicated by †q < 0.05 (Fisher’s exact test and a FDR using Benjamini-Hochberg method).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/18/10.1182_bloodadvances.2022008355/5/m_advancesadv2022008355f2.png?Expires=1767710556&Signature=tlLdefGs5GcM1gS-VaBe4QT5Bi8974U-cUMr4JmyI400cbTeYacK8QHRN4kID4XPJpT~bg2yyNgqhNlqs6qAVZqpBLMGRGJJ9m3oUW0ViKGbflCUkLeVUzFOlkO55xChO~EU~AE0i4rqq9HC6nGEwelH7ac2P~Au6hjJgBNKKH60MOlDQlsZYW5qbcGBiXqWaTJwRlMGelrygFn4oKD831rtq43mObF81jm~eqpoZ8q31VsSUG~wq-Dxh05OsJp~mQzMLmnQwVN2jY2kNzvP70w4RGJn47pELFuVYImG0ulzRbNg6hu1xPVlYmgVu2fkot7iFqE-tp6TwkzdLc6anw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Microenvironment, mutations, translocations, and copy number landscape of stage I vs stage III/IV FL. (A) Percentage of positive nucleated cells for CD3, CD4, CD8, FOXP3, and PD1 depicted as boxplots. For CD163 and CD68, the percentage of positive area of the total cell area was computer-assisted scored are plotted in the boxplots (stage I in red [n = 73] and stage in blue III/IV [n = 120]). Significant differences are seen for CD8, PD1, CD163, and CD68 with a P value adjusted for multiple testing: *P = .02, **P = .002, ***P < .001. (B) Comparison plots for CNAs between stage I as filled areas (n = 82) and stage III/IV as lines (n = 139) are percentages of the number of cases with gains (positive value, blue) and losses (negative value, red), sorted for chromosome position (x-axis). (C) Frequency plots with P values (blue) calculated with a 2-sided rank-sum test with 10 000 permutations and FDR (striped segments) of the difference in CNAs; the horizontal dotted lines show the significance threshold P values <.05 in blue, and the FDR <0.1 in red. If the difference in CNA level crosses the P value, and the FDR level is <0.1, the difference is considered significant, which is indicated by * 6q23.3 and 9p21-22 (*P < .05). (D) Frequency of BLC2 and BLC6 translocations and top 20 mutated genes according to stage I in red (n = 82) and stage III/IV in blue (n = 139); significant differences are indicated by †q < 0.05 (Fisher’s exact test and a FDR using Benjamini-Hochberg method).
Microenvironment, mutations, translocations, and copy number landscape of stage I vs stage III/IV FL. (A) Percentage of positive nucleated cells for CD3, CD4, CD8, FOXP3, and PD1 depicted as boxplots. For CD163 and CD68, the percentage of positive area of the total cell area was computer-assisted scored are plotted in the boxplots (stage I in red [n = 73] and stage in blue III/IV [n = 120]). Significant differences are seen for CD8, PD1, CD163, and CD68 with a P value adjusted for multiple testing: *P = .02, **P = .002, ***P < .001. (B) Comparison plots for CNAs between stage I as filled areas (n = 82) and stage III/IV as lines (n = 139) are percentages of the number of cases with gains (positive value, blue) and losses (negative value, red), sorted for chromosome position (x-axis). (C) Frequency plots with P values (blue) calculated with a 2-sided rank-sum test with 10 000 permutations and FDR (striped segments) of the difference in CNAs; the horizontal dotted lines show the significance threshold P values <.05 in blue, and the FDR <0.1 in red. If the difference in CNA level crosses the P value, and the FDR level is <0.1, the difference is considered significant, which is indicated by * 6q23.3 and 9p21-22 (*P < .05). (D) Frequency of BLC2 and BLC6 translocations and top 20 mutated genes according to stage I in red (n = 82) and stage III/IV in blue (n = 139); significant differences are indicated by †q < 0.05 (Fisher’s exact test and a FDR using Benjamini-Hochberg method).
Genomic and microenvironmental features of stage I FL compared with stage III/IV
BCL2trls were detected with significantly lower frequency in stage I cases; that is, 59% compared with 91% of cases in stage III/IV disease (P < .001) (Figure 2B). There were no differences in the breakpoint locations between the stages (supplemental Figure 2A-F; supplemental Table 9). In addition to classical BCL2/IGH translocations (n = 171), rare other translocation partners were found: IGL (n = 2), IGK (n = 1), and HLA-DRA (n = 1), all present in stage III/IV.
BCL6trls were observed in 6% of stage I cases and 17% of stage III/IV cases (P = .07) (Figure 2B). Translocation partners for BCL6 were diverse (supplemental Figure 2G-H; supplemental Table 9).
Other IGH translocations were observed in 13% of stage I cases and 16% of stage III/IV cases, with diverse translocation partners. In addition, most recurrent other translocations detected were MYC (stage I, n = 1; stage III/IV, n = 3) and TBL1XR1 (stage I, n = 1; stage III/IV, n = 3) translocations (supplemental Table 9).
High-quality genome-wide CNA plots were obtained by shallow whole-genome sequencing for all cases. Overall, stage I and III/IV disease displayed comparable frequencies of aberrations, and the spectrum of alterations did not differ significantly (Figure 2B; supplemental Table 10). The copy number load per stage is similar (supplemental Figure 3A). The overall landscape of CNA included focal gains of known FL-related genes such as REL and BCL11A (2p16.1) and focal losses of TNFRSF14 (1p36.32), PRDM1 (6q21), TNFAIP3 (6q23.3), CDKN2A (9p21-22), and PTEN and FAS (10q23.31). The focal loss of 9p21-22 containing CDKN2A, and a small region on 6q12 without a specific gene, were significantly more common in stage III/IV (Figure 2C).
The median number of nonsynonymous and splice-site mutations was comparable between stage I (median, 11 mutations per case; range, 0-29) and stage III/IV (median, 12 mutations per case; range, 0-116) (supplemental Figure 3B; supplemental Table 11). Regarding BCL2 somatic hypermutations (SHMs) (defined for the purpose of this study as ≤2 mutations in known SHM target genes), there was a significant difference between the number of cases with SHMs in stage I (36%) and stage III/IV (71%; P = .017), which correlated with more BCL2trls in stage III/IV. Comparing the number of SHM-related mutations only in the BCL2trl+ cases, there was no difference (stage I 63% [n = 26 of 41] vs stage III/IV 77% [n = 97 of 127]; P = 1). SHM in BCL6 (stage I, n = 1) and PIM1 (stage I, n = 4; stage III/IV, n = 3) was seen in only a few cases, and no SHM was found in MYC (supplemental Table 11).
Of the genes included in the LLBC-NGS targeted panel, the following were most frequently affected by nonsynonymous mutations in stage I FL: KMT2D (52%), CREBBP (50%), BCL2 (35%), EZH2 (35%), TNFRSF14 (35%), STAT6 (30%), and MEF2B (18%). The most frequently affected genes in stage III/IV FL were: KMT2D (76%), CREBBP (69%), BCL2 (54%), TNFRFS14 (31%), EZH2 (20%), and ARID1A (17%). A comparison of mutation frequencies found that STAT6 was mutated at a significantly higher frequency in stage I compared with stage III/IV (FDR <0.001), whereas KMT2D and CREBBP were mutated at a significantly higher frequency in stage III/IV malignancies (FDR = 0.003 and FDR = 0.04) (Figure 2D).
Overall, the mutational landscape between stage I FL and stage III/IV FL was highly similar, with a dominant involvement of epigenetic and chromatin-modifying genes (KMT2D, CREBBP, EP300, EZH2, and MEF2B) but at different frequencies. In 94% of stage I cases and in 99% of stage III/IV cases, at least 1 of these 5 genes was mutated, indicating a critical role of epigenetic deregulation in the development of FL.
Integrated analysis of translocation, CNA, and mutation data
Next, we performed an integrated analysis of all molecular modalities using an unsupervised hierarchical clustering strategy to explore the potential heterogeneity and integrated profiles within stage I disease (Figure 3). For this analysis, 81 stage I cases were included; 1 case with a very low level of shared mutations was excluded. The Dunn index estimates 4 clusters for stage I as optimal (supplemental Figure 4).
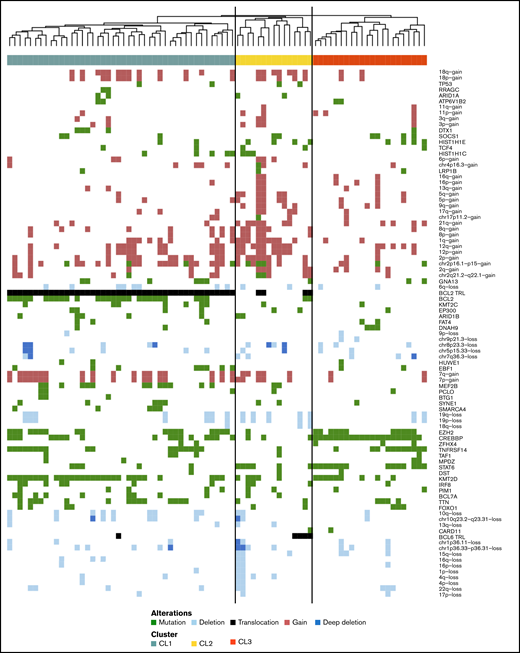
Hierarchic cluster analysis of stage I FL. Features of stage I (n = 81) included in unsupervised hierarchical clustering are somatic mutations present in >5% of the cases, BCL2 and BCL6 translocations, and focal and chromosomal arm–level aberrations present in >5% of the samples with Spearman correlation. Each column represents one patient, CL1 (green, n = 44), CL2 (yellow, n = 15) and CL3 (orange, n = 22). Mutations (green), translocations (black) and CNAs (gains red, losses light blue and multiple losses dark blue) are ordered in the rows.
Hierarchic cluster analysis of stage I FL. Features of stage I (n = 81) included in unsupervised hierarchical clustering are somatic mutations present in >5% of the cases, BCL2 and BCL6 translocations, and focal and chromosomal arm–level aberrations present in >5% of the samples with Spearman correlation. Each column represents one patient, CL1 (green, n = 44), CL2 (yellow, n = 15) and CL3 (orange, n = 22). Mutations (green), translocations (black) and CNAs (gains red, losses light blue and multiple losses dark blue) are ordered in the rows.
Cluster 1 (CL1) (n = 44) was characterized by presence of BCL2trl in all cases in concert with frequent mutations in BCL2. CL1 was further characterized by classic FL mutations (KMT2D, EZH2, CREBBP, TNFRSF14, and MEF2B) (supplemental Figure 5; supplemental Table 12). Cluster 2 (CL2) (n = 15) was characterized by a relatively high level of CNAs (median, 19%; range, 0%-58%) (supplemental Figure 6) and mutations in 40% of the cases in one or both linker histone genes (HIST1H1E, 27%; HIST1H1C, 20%). In CL2, BCL2trl and BCL6trl and STAT6 mutations occurred at intermediate frequencies, whereas epigenetic modifying genes (KMT2D, CREBBP, and EZH2) were mutated at relatively low levels (supplemental Table 12). The last 2 clusters were defined by the absence of BCL2trl and presence of STAT6 and CREBBP mutation. They differ in presence of TNFRSF14 and KMT2D mutations, which are both tumor suppressor genes. TNFRSF14 is controlled by KMT2D51 ; the biological pathways of these two clusters are likely identical, and thus we combined these two clusters into cluster 3 (CL3) (n = 22). CL3 has the “classical” FL-related genes with the exception of MEF2B (Figure 3; supplemental Figure 7; supplemental Table 12). The mean consensus index of 2 samples from the same cluster was 87%, indicating that the clustering was stable (supplemental Figure 8).
Integrated analysis of the stage III/IV FL cases revealed BCL2trl as the most frequent genetic alteration, resulting in a relatively homogeneous cluster of BCL2trl+ FL (n = 128) and a separate cluster lacking the BCL2trl and concomitant BCL2 mutations (n = 11) (supplemental Figures 7, 9, and 10). In this BCL2trl-negative (BLC2trl–) group, BCL6trl were present at high frequency (64%). Mostly, classical FL mutations were seen, albeit at different frequencies for BCL2trl+ vs BCL2trl–cohort (KMT2D, 79% vs 55%; EZH2, 22% vs 0%; HIST1H1E, 15% vs 0%; and HIST1H1C, 6% vs 27%) (supplemental Table 12).
The stage III/IV BCL2trl– cluster lacked the distinct characteristics of stage I BCL2trl– CL3 and was not enriched for CREBBP and STAT6 mutations; in addition, characteristic high-level CNAs of CL2 were less prominent. Altogether, the results showed that within stage I, there are 2 distinct molecularly driven clusters in addition to a “canonical” FL cluster.
CNAs, mutations, and translocations of the subset with complete microenvironment information were representative of the data set with complete NGS. The clustering would not have been affected if this subset was used to perform the analysis (supplemental Figures 11-13).
After the identification of the 3 clusters in stage I disease, we explored whether these clusters might underlie a distinct immune microenvironment signature. For 183 of 220 cases included in the hierarchical cluster analysis, complete microenvironment information was available for an integrated analysis (stage I, n = 69: CL1 n = 37, CL2 n = 11, CL3 n = 21; and stage III/IV, n = 114: BCL2trl+ n = 107, BCL2trl– n = 7) (supplemental Figure 14; supplemental Table 13). Although there seemed to be a lower level of PD1+ cells in CL2, due to the few cases per cluster and the minimal differences observed in the scoring results, no statistical analysis could be performed, which precluded biological interpretation of the data.
Discussion
Tumor and microenvironmental analyses of the largest series of nodal stage I FL thus far allow us to conclude that stage I FL has mostly genomic and microenvironmental abnormalities similar to those of stage III/IV disease. However, some significant differences were found.
In both groups, the same mutations and CNAs were observed, with KMT2D, CREBBP, BCL2, TNFRSF14, and EZH2 as the most frequently mutated genes. The most frequent copy number gains of chromosomes 1q, 2, 7, 12, and 18 in the present series are in agreement with other published reports.52-58 The immunophenotype of the tumor cells is also consistent with germinal center cell derivation, and the overall composition of the immune microenvironment shows no clinically significant differences between stage I and stage III/IV disease. The higher frequency of CD8+ T cells, lower frequency of PD1+ T cells, and CD68/CD163+ macrophages noted in stage I FL are suggestive of a biological role for these cell populations; however, the small absolute differences cannot be appreciated without automated image analysis and are therefore unlikely to be useful in clinical practice. Thus, our results are consistent with the currently accepted view that stage I FL in general is not a distinct biological entity.
The major difference observed between stage I and stage III/IV is the frequency of BCL2trl, as previously reported.15 Studies focusing on BCL2trl– FL have reported a higher frequency of BCL6trl,59 as well as more frequent mutations in STAT6, CREBBP, and TNFRSF14.60-63 Our data allow a broader perspective as we now identify signatures in their specific clinical contexts showing essentially different signatures for BCL2trl– stage I and stage III/IV disease. A unique BCL2trl– cluster, CL3, is recognized as highly specific for stage I FL. CL3 is characterized by enrichment for CREBBP (95%), STAT6 (64%), EZH2 (50%), and TNFRSF14 (50%) mutations and absence of BCL6trl, whereas stage III/IV BCL2trl– FL is enriched for BCL6trl (64%) with low frequency of the most frequently mutated genes in CL3. These differences suggest that different sets of specific molecular events may drive the pathogenesis of FL.
We identified the same 3 most frequent hotspots in STAT6 previously reported by Yildiz et al.64 E372K (stage I, n = 5; stage III/IV, n = 1), E377K (stage I, n = 3; stage III/IV, n = 3), and D419G (stage I, n = 5; stage III/IV, n = 2) (supplemental Figure 15A-B), which are activating mutations in the interleukin-4 (IL4)/JAK/STAT pathway. This pathway may indeed be capable of overriding the important role of the absent BCL2trl in the pathogenesis of stage I FL. For example, follicular T-helper cells are an important source of IL4, which can directly regulate BCL2 expression via STAT6.65 Due to the low number of cases in each of the 3 clusters and minimal differences in frequency of PD1+ follicular T-helper cells in the microenvironment, we are not yet able to draw firm conclusions about the interaction between STAT6 mutations and the number of PD1+ follicular T-helper cells, however. Strikingly, there is only 1 BCL2trl– sample with a STAT6 mutation in the stage III/IV group.
The CL2 cluster appears to represent a distinct group defined by a higher number of CNAs and higher frequency of HIST1H1E and HIST1H1C mutations (supplemental Figure 15C-E). Loss of function of these linker histone genes has been shown to drive lymphomagenesis due to higher fitness of germinal center B cells and enhanced self-renewal potential.66 The small number of cases in this cluster and the relatively heterogeneous features, however, preclude definite interpretation.
It should be noted that although clustering is supported by mathematical and biological evidence, it should not be regarded as a definitive classification but rather a means to obtain biological insight and a step toward finding the driver genes and pathways of FL.
A limitation in our study is that the majority of patients were diagnosed before including fluorodeoxyglucose–positron emission tomography as a staging procedure, and therefore, this cohort may contain some patients who would be classified as a higher stage with current staging techniques. As indicated in the literature, up to 30% of patients may be upgraded to a higher stage with positron emission tomography/computed tomography scans.67 However, the unique and specific mutational landscape characteristics of the 2 distinct clusters described here are not recognized in the advanced stage, suggesting that the majority of these cases were true stage I FL.
The identification of 3 different clusters raises the question if each subtype follows a distinct clinical course. Due to the diverse origin of the samples, treatment modalities, follow-up strategies, and the limited number of samples per cluster, our study is not able to address answers with regard to clinical outcome per cluster.
In conclusion, with this relatively large cohort, we showed that stage I FL is a genetically heterogeneous group divided over 3 distinct and unique clusters, for which the two BCL2trl– clusters suggest different underlying oncogenetic pathways compared with stage III/IV FL. Our results suggest that BCL2trl– stage I disease follows a different pathogenesis than BCL2trl– stage III/IV.
Acknowledgments
The authors thank all pathologists and pathology laboratories for providing tissue materials and data about patients who have been under their care, the EORTC for permission to use the data from EORTC study 20971/22997 for this research, Yongsoo Kim and Jurriaan Janssen for advice on cluster analysis, and Anton Hagenbeek, the founding father of the Lunenberg Lymphoma Biomarker Consortium. They also thank the Hartwig Medical Foundation for generating, analyzing, and providing access to reference the whole-genome sequencing data of The Netherlands population.
This study was supported by the Dutch Cancer Society (grant KWF 2015-7925) and by unrestricted grants from van Vlissingen Lymfoom Fonds, Genentech/Roche, GlaxoSmithKline, Pfizer Pharma, Teva Pharmaceuticals/Cephalon, Millenium Pharmaceuticals Inc., and Celgene. The HMRN is funded by Cancer Research UK (grant 29685) and Blood Cancer UK (grant 1503).
G.T.L.-d.V., D.d.J., B.Y., and M.J.K. received funding from the Dutch Cancer Society (grant KWF 2015-7925). W.B.C.S., E.v.D., C.L.-J., and D.M.-J. were funded by unrestricted grants from Vlissingen Lymfoom Fonds, Genentech/Roche, GlaxoSmithKline, Pfizer Pharma, Teva Pharmaceuticals/Cephalon, Millenium Pharmaceuticals Inc., and Celgene. A.G.S. was funded by Malignancy Research Network is funded by Cancer Research UK, grant numbers 29685; and Blood Cancer UK, grant number 1503.
Authorship
Contribution: The LLBC, M.J.K., B.Y., and D.d.J. designed the study; G.T.L.-d.V., W.B.C.S., E.v.D., C.L.-J., A.J.C., P.S., M.M., and N.J.H. performed experiments; G.T.L.-d.V., W.B.C.S., E.v.D., C.L.-J., M.G.M.R, M.M., B.S., A.R., D.M.-B., B.Y., and D.d.J. analyzed and interpreted the data; G.T.L.-d.V., W.B.C.S, E.v.D., D.M.-B., C.L.-J., B.Y., and D.d.J. wrote the manuscript; and all authors critically revised the manuscript, were involved in its editing, and gave final approval of the submitted and published versions.
The LLBC is a collaboration of 9 international lymphoma research groups, each represented by a clinical investigator and one or more hematopathologists and supported by a team of statisticians. Foundation of the LLBC was made possible with a grant from the Van Vlissingen Lymphoma Foundation. EORTC Lymphoma group represented by: Daphne de Jong and John Raemaekers. HOVON Lymphoma group represented by: Daphne de Jong and Marie-José Kersten. Lymphoma Study Association represented by: Philippe Gaulard, Gilles Salles, Luc Xerri, Delphine Maucort-Boulch, and Carole Langois-Jacques. British Columbia Cancer Agency represented by: Laurie H. Sehn and David W. Scott. GLSG represented by: Andreas Rosenwald, Wolfram Klapper, Christian Buske, Wolfgang Hiddemann, Eva Hoster, and Heike Horn. Nordic lymphoma group represented by: Birgitta Sander and Eva Kimby. Barts Cancer Institute represented by: Maria Calaminici, John Gribben, and Andrew J. Clear. HMRN represented by: Catherine Burton, Reuben M. Tooze, and Alexandra G. Smith. Stanford represented by: Yasodha Natkunam and Ranjana Advani.
Conflict: None of the authors has a relevant conflict of interest.
Correspondence: Wendy B. C. Stevens, Geert Grooteplein Zuid 8, Nijmegen, 6525 GA, The Netherlands; e-mail: wendy.stevens@radboudumc.nl.

