

Key Points
Stem cell transplant improves long-term survival in T/NK CAEBV, though mortality remains high.
Development of T/NK lymphoma showed a trend with increased mortality.

Abstract
Chronic active Epstein-Barr virus (EBV) disease (CAEBV) is characterized by high levels of EBV predominantly in T and/or natural killer cells with lymphoproliferation, organ failure due to infiltration of tissues with virus-infected cells, hemophagocytic lymphohistiocytosis, and/or lymphoma. The disease is more common in Asia than in the United States and Europe. Although allogeneic hematopoietic stem cell transplantation (HSCT) is considered the only curative therapy for CAEBV, its efficacy and the best treatment modality to reduce disease severity prior to HSCT is unknown. Here, we retrospectively assessed an international cohort of 57 patients outside of Asia. Treatment of the disease varied widely, although most patients ultimately proceeded to HSCT. Though patients undergoing HSCT had better survival than those who did not (55% vs 25%, P < .01), there was still a high rate of death in both groups. Mortality was largely not affected by age, ethnicity, cell-type involvement, or disease complications, but development of lymphoma showed a trend with increased mortality (56% vs 35%, P = .1). The overwhelming majority (75%) of patients who died after HSCT succumbed to relapsed disease. CAEBV remains challenging to treat when advanced disease is present. Outcomes would likely improve with better disease control strategies, earlier referral for HSCT, and close follow-up after HSCT including aggressive management of rising EBV DNA levels in the blood.
Introduction
Approximately 95% of adults worldwide have been infected with Epstein-Barr virus (EBV).1 Following initial infection, the virus establishes latency in B cells. In rare individuals with chronic active EBV (CAEBV), EBV is predominantly found in T and/or natural killer (NK) cells, with persistently high levels of EBV in the blood and tissues.2 Historically, these cases have been more common in persons of Asian and South/Central American descent.3 The hallmark of disease is proliferation of EBV-infected T or NK cells that infiltrate tissues, leading to end-organ damage. Patients present with fevers, hepatosplenomegaly, liver inflammation, cytopenias, and lymphoproliferation.4 In more aggressive versions of the disease, hemophagocytic lymphohistiocytosis (HLH), liver failure, and progression to NK/T-cell lymphomas may occur; these are the most common causes of death.5 The disease becomes extremely difficult to treat once it has progressed to significant organ dysfunction, particularly severe hepatic impairment. Although several germline genetic defects are known to cause susceptibility to severe EBV disease,6 and some patients with CAEBV have somatic mutations in their T or NK cells,7 a causative monogenetic defect has not been identified in most patients with CAEBV.
Treatment considerations remain challenging for patients with CAEBV. Expert consensus is to use corticosteroids and/or ganciclovir in combination with bortezomib or histone deacetylase inhibitors, as well as cytotoxic chemotherapy for those with fulminant presentation to reduce disease burden and improve the patient’s condition prior to allogeneic hematopoietic stem cell transplantation (HSCT).8 In the largest review of patients with CAEBV in the United States,9 long-term survival was poor at ∼30%, although only ∼40% underwent HSCT. Most patients died due to progression of their disease despite treatment. Survival was improved in those patients who were younger, had shorter duration of disease, and were referred early for HSCT. Of 8 patients that underwent HSCT, 5 survived. Three of these 5 surviving patients received chemotherapy before HSCT, and 2 of the survivors received EBV-specific cytotoxic T cells after HSCT. These reported outcomes are similar to contemporary cohorts of CAEBV in the Asian population, with 3-year survival near 60%.10 The optimal treatment to decrease EBV burden in these patients before transplantation, as well as how to best condition patients for transplant, is currently unknown.
Our primary aim was to describe a contemporary cohort of CAEBV patients outside Asia, focusing on demographic and clinical characteristics, including age, presenting symptoms, treatment modalities, responses, and outcome. All but 1 of these cases are previously unreported; thus, they provide an updated analysis on the management of CAEBV since the prior study of patients in the United States from 1982 to 2010.9 We hypothesized that patients undergoing HSCT with adequate disease control, as defined by fewer systemic signs and a low viral load immediately before their preparative regimen, would have the best outcomes.
Methods
Patients
Patients with CAEBV were defined by disease persisting ≥3 months, persistently elevated EBV in the blood by DNA polymerase chain reaction (PCR), systemic inflammatory symptoms, infiltration of tissues with EBV-infected T or NK cells, and no known immunodeficiency. Identification of the cell type containing EBV was determined by confirming which cell types (using CD3 or CD56 antibody staining) were EBV-encoded RNA-positive by in situ hybridization of tissue or, in ambiguous cases, by sorting peripheral blood into populations of B, T, and NK cells and determining which population had the highest level of EBV DNA. EBV DNA PCR results were reported as in copies/mL in whole blood. Most patients were referred after their physicians requested information for treatment in an anti-EBV cellular therapy protocol [Cytotoxic T Cells to Treat Relapsed EBV-positive Lymphoma, ClinicalTrials.gov Identifier: NCT01956084] and/or enrolled in a CAEBV protocol [Genetics of CAEBV, ClinicalTrials.gov Identifier: NCT00032513] at the National Institutes of Health (NIH). Patients presented with CAEBV between 2009 and 2020. With 1 exception, none of the patients were reported previously; all were diagnosed outside of Asia. All collected data were deidentified, captured, and summarized by the host institution. Data use analysis agreements were obtained for organizations that required them, and Institutional Review Board approval was obtained from the Children’s National Hospital per institutional guidelines. Genomic analysis was not required for this study; however, familial HLH was excluded for patients presenting with this complication. Patients from the NIH cohort signed informed consent on a protocol approved by the NIH Institutional Review Board.
Statistical analysis and definitions
Patient characteristics included country of origin, age at diagnosis, sex, and race/ethnicity. Disease aspects included cell-type involvement (NK and/or T), viral load at diagnosis, presence of cytopenia, diagnosis of secondary HLH, and other complications. Patients with B-cell EBV–mediated lymphoproliferative disease and familial HLH were excluded. Treatments used before HSCT and responses to HSCT were recorded. For patients that proceeded to HSCT, viral load prior to HSCT, donor and preparative regimen, as well as outcomes, were obtained.
Ethnicity categories were classified as white (non-Hispanic), Asian, Native American, and Hispanic. Partial responses were defined as a reduction of ≥1 log in viral load as previously published.11 Viral load before HSCT was divided between low level, defined as <10 000 copies/mL, or increased level, defined as ≥10 000 copies/mL. These values were chosen because they were felt sufficiently separate from a high viral burden, previously defined as 100 000 copies/mL.11 Full responses were defined as low-level or no viremia with resolution of all disease manifestations. Reduced intensity conditioning (RIC) and myeloablative conditioning (MAC) regimens were categorized as per the Center for International Blood and Marrow Transplant Research definitions.12 As HSCT is considered curative, patients post-HSCT were deemed in complete remission if they had resolution of disease and no further lymphoproliferation.
The log-rank test was used to investigate the association between survival time and the following variables: ethnicity, age at presentation, cell type involved (T vs NK), diagnosis of HLH, use of chemotherapy, development of lymphoma, use of HSCT, viral load before HSCT, use of serotherapy, use of radiation, and preparative regimen used (RIC vs MAC). These were then recalculated using a Cox proportional hazards model.
Results
Patient characteristics
A total of 57 patients were included in this study (Table 1). Most patients (93%) were diagnosed in the United States; others were identified in Canada, Israel, and New Zealand. All patients except for patient #2113 are previously unreported, and all but 1 was diagnosed in the last decade (2010-2020). The mean age at diagnosis was 13.6 years (range 1.8-37). Although 74% (42/57) of patients were less than 18 years old, most (28/42, 67%) were ≥8 years old at the time of diagnosis. There was no sex predilection, with 56% (32/57) of cases occurring in females. Most (49% [28/57]) cases occurred in patients of Hispanic and/or Asian ancestry as previously described. This included 18 patients of Hispanic descent and 10 of Asian ancestry. However, a third (22/57) of enrolled subjects identified as white, and 12% (7/57) were Native American.
Summary of patients based on transplant type
| Type of HSCT | Number of patients | Mean Age (yrs) at diagnosis (range) | Gender | Ethnicity | Cell line affected | Number of patients with lymphoma | Outcome |
|---|---|---|---|---|---|---|---|
| RIC | 27 | 7.1 (0.8-36) | 15 M | 11 H | 20 T | 9 | 12 died |
| 12 F | 8 W | 5 T, NK | 14 alive | ||||
| 5 A | 2 NK | 1 other | |||||
| 2 NA | |||||||
| 1 M | |||||||
| MAC | 13 | 15.5 (5-37) | 4 M | 5 W | 7 T | 7 | 5 died |
| 9 F | 4 NA | 5 NK | 8 alive | ||||
| 3 H | 1 T, NK | ||||||
| 1 A | |||||||
| Sequential: MAC/RIC or RIC/MAC | 3 | 11.3 (1.8-21) | 1 M 2 F | 1 W 1 NA 1 M | 3 T | 2 | 1 died 2 alive |
| Unknown/not infused HSCT | 6 | 12.7 (2.4-32) | 2 M 4 F | 3 W 2 H 1 A | 4 T 1 NK 1 T/NK | 1 | 5 died 1 other |
| No HSCT | 8 | 13.7 (2-25) | 3 M 5 F | 5 W 2 H 1 A | 5 T 2 NK 1 T, NK | 4 | 6 died 2 alive |
| Total | 57 | 13.6 (1.8-37) | 25 M 32 F | 22 W 18 H 8 A 7 NA 2 M | 39 T 10 NK 8 Both | 23 | 29 died 26 alive 2 other |
| Type of HSCT | Number of patients | Mean Age (yrs) at diagnosis (range) | Gender | Ethnicity | Cell line affected | Number of patients with lymphoma | Outcome |
|---|---|---|---|---|---|---|---|
| RIC | 27 | 7.1 (0.8-36) | 15 M | 11 H | 20 T | 9 | 12 died |
| 12 F | 8 W | 5 T, NK | 14 alive | ||||
| 5 A | 2 NK | 1 other | |||||
| 2 NA | |||||||
| 1 M | |||||||
| MAC | 13 | 15.5 (5-37) | 4 M | 5 W | 7 T | 7 | 5 died |
| 9 F | 4 NA | 5 NK | 8 alive | ||||
| 3 H | 1 T, NK | ||||||
| 1 A | |||||||
| Sequential: MAC/RIC or RIC/MAC | 3 | 11.3 (1.8-21) | 1 M 2 F | 1 W 1 NA 1 M | 3 T | 2 | 1 died 2 alive |
| Unknown/not infused HSCT | 6 | 12.7 (2.4-32) | 2 M 4 F | 3 W 2 H 1 A | 4 T 1 NK 1 T/NK | 1 | 5 died 1 other |
| No HSCT | 8 | 13.7 (2-25) | 3 M 5 F | 5 W 2 H 1 A | 5 T 2 NK 1 T, NK | 4 | 6 died 2 alive |
| Total | 57 | 13.6 (1.8-37) | 25 M 32 F | 22 W 18 H 8 A 7 NA 2 M | 39 T 10 NK 8 Both | 23 | 29 died 26 alive 2 other |
Yrs, years; H, Hispanic; W, White; A, Asian; NA, Native American; M, mixed.
EBV results and disease-related complications
Sixty-five percent (37/57) of subjects had T-cell CAEBV, 18% (10/57) had NK cell disease, 14% (8/57) had NK and T-cell disease, and 4% (2/57) had T- and B-cell disease. The average EBV load in whole blood at diagnosis was 5.7 million copies/mL (median 630 957 copies/mL).
Cytopenias were the most common presenting sign, present in 80% (41/51) of patients (Figure 1). Anemia and thrombocytopenia were equally common in the cohort (63% [32/51]). Multilineage cytopenias were frequently encountered; 69% of all patients (35/51) had more than 1 cell line decreased at presentation.
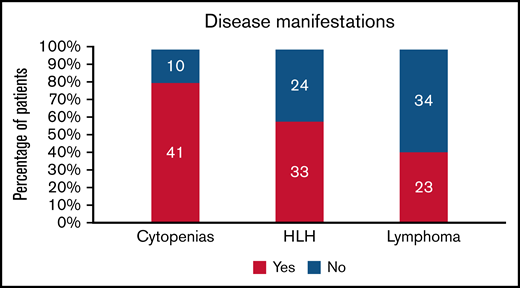
Disease manifestations in patients with CAEBV. The number of patients is shown in the bars, and the percentage is on the y-axis.
Disease manifestations in patients with CAEBV. The number of patients is shown in the bars, and the percentage is on the y-axis.
Two-thirds of patients (34/52) had hepatic impairment, including elevation in liver function enzymes and/or impaired synthetic function. Over half (33/57) met HLH criteria14,15 in their disease course. They were managed with standard treatments, including dexamethasone and etoposide. Relapsed/refractory HLH was common in the cohort; 27% (9/33) of those who developed this complication ultimately died of it.
Forty percent of patients (23/57) developed NK and/or T-cell lymphoma during their course; 11 (19% of cohort) had lymphoma diagnosed while being monitored for their CAEBV symptoms and before any treatment. Two patients presented as hydroa vacciniforme–like lymphoproliferative disease (HVLPD) before developing systemic symptoms. At least 2 patients had vascular involvement, including coronary and cerebral artery aneurysms and lymphocytic myocarditis.
Management before HSCT
Several patients were monitored before any intervention. Their course was quite varied, but many were undiagnosed before expert review; 1 subject received his first treatment 3 years after onset of symptoms. Treatment was varied before HSCT, and no consistent approach was used (Figure 2; supplemental Table 1). Nearly half (25/57) of patients received rituximab. As would be expected for a NK/T-cell–mediated disorder, only a minority of patients (32% [8 of 25]) had a partial response; there were no sustained or complete responses.
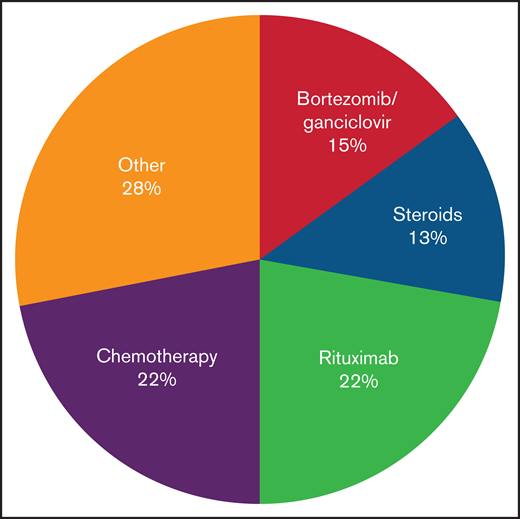
Treatments received in patients with CAEBV before HSCT. Treatment approaches varied; the most common approaches included chemotherapy and combination bortezomib/ganciclovir.
Treatments received in patients with CAEBV before HSCT. Treatment approaches varied; the most common approaches included chemotherapy and combination bortezomib/ganciclovir.
Combination bortezomib with ganciclovir is a potential therapy for disease control. Bortezomib can induce lytic EBV gene expression, including a viral protein kinase which phosphorylates ganciclovir, resulting in cell death.8 Thirty percent (17/57) of patients received this combination, resulting in a reduction of EBV loads in 71% (12 of 17) of patients; however, the responses were not sustained in most cases. It has been our experience that patients with active secondary HLH were less likely to respond to bortezomib/ganciclovir, but this could not be quantified in this survey.
Previous reports have shown that patients with CAEBV are not likely to respond to autologous EBV-specific T-cell therapy.16 This is thought to be due to an intrinsic impairment in the function of the patient’s T-cells. In this cohort, only 1 received these cells before HSCT; there was no response.
Chemotherapy was administered to those refractory to the above interventions, including those with lymphoma. Most regimens followed were well-described NK and/or T-cell lymphoma treatments, including anthracycline-based regimens (EPOCH, CHOP), or l-asparaginase including regimens such as SMILE17 (data not shown). Most patients (84% [21/25]) had improvement in symptoms as well as significant reductions in viral load. Ultimately, this proved the most effective way to reduce EBV-related complications before HSCT and was used in almost half of all cases. In at least 3 cases, chemotherapy was planned but could not be given due to organ dysfunction and/or poor functional status.
Other treatments tried include romidepsin, sirolimus, vorinostat, hydroxychloroquine, thalidomide, azathioprine, and colchicine (Figure 2; supplemental Table 1). Seven patients received 1 or more of these therapies; there were no responses. Immunomodulatory drugs such as etanercept and anakinra were also used in 3 patients; although use of these medications did not result in EBV load reduction, they did help control manifestations of HLH in 2 patients. In 1 patient with a clonal population of CD30+ T cells, brentuximab-vedotin resulted in a complete response; the patient subsequently proceeded to HSCT.
Recently, attempts have been made to directly block T-cell stimulation and/or proliferation in patients with CAEBV. Three patients in our cohort received alemtuzumab and had remission of their disease, though not sustained. The authors are aware of other patients with CAEBV, not in this cohort, whose disease rapidly worsened with alemtuzumab, likely due to further loss of T-cell control of EBV-infected cells. Two patients in our cohort received ruxolitinib; 1 had a prolonged partial remission with this therapy alone. One patient received nivolumab with a complete response.
HSCT donors and conditioning regimens
Eighty-six percent (49/57) of patients proceeded to HSCT as definitive therapy for their disease (supplemental Table 2). Sixty-one percent (30/49) of patients underwent preparative regimens classified as RIC (Table 1; supplemental Table 2). Thirty-nine percent (19/49) of the regimens included total body irradiation. There were no significant differences in survival rates with RIC vs MAC regimens, radiation-based vs chemotherapy-only regimens, or use of serotherapy (alemtuzumab/antithymocyte globulin vs none).
Forty-nine donors were used for 46 transplants; 3 patients started conditioning but died before donor infusion. A similar proportion of related (27/49; 55%) and unrelated (21/49; 45%) donors as well as matched (25/49; 51%) and mismatched (22/49; 45%) donors was noted; 12 (24%) were matched related donors. Most donors, including all related donors, were evaluated for EBV seropositivity and whole blood EBV DNA PCR. Ninety-seven percent (35/36) of evaluated donors were EBV+. The related donor of patient 31, who was EBV-seropositive, subsequently developed an NK/T-cell lymphoma, raising concern for a genetic predisposition. The recipient subsequently relapsed and required a second HSCT from an unrelated donor. In total, 3 patients (6% of those undergoing HSCT) underwent more than 1 transplant. Two patients received autologous transplants for treatment of lymphoma before HSCT.
Outcome
With 2 patients lost to follow-up, the survival rate was 47% (26/55) at a median and mean follow-up of 18 months and 24 months, respectively, from the time of diagnosis. Three patients who planned to receive HSCT died during their conditioning regimen and before stem cell infusion; therefore, they were not included in the HSCT group analysis. Survival for those undergoing HSCT was significantly (P < .01) higher; 55% (24/44) compared with 25% (2/8) for those who did not have a transplant (Figure 3). Median survival for the HSCT group was 52 months compared with 12.5 months for those not transplanted. Two of the 3 patients requiring a second HSCT were alive at last follow-up. Ethnicity, age at presentation, HLH as a complication, and the use of chemotherapy pre-HSCT were not found to confer a higher risk of survival or death.
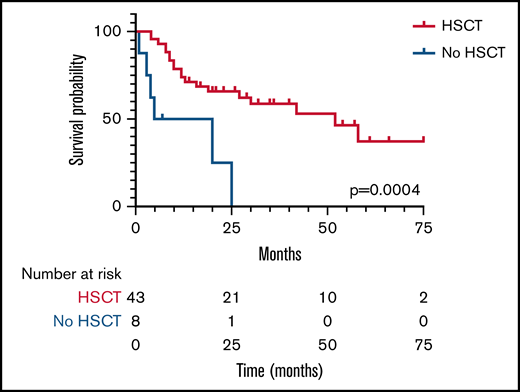
Kaplan-Meier plot of survival in patients undergoing HSCT (blue line) or no HSCT (red line). Median survival time was 52 months in the HSCT group and 12.5 months in the no-HSCT group. The number of patients still evaluable at different times after HSCT is shown below the graph.
Kaplan-Meier plot of survival in patients undergoing HSCT (blue line) or no HSCT (red line). Median survival time was 52 months in the HSCT group and 12.5 months in the no-HSCT group. The number of patients still evaluable at different times after HSCT is shown below the graph.
Of 23 patients who developed lymphoma, 8 (35%) are currently alive. Survival was higher in those who did not develop lymphoma in their course (56% vs 35%, P = .11). All patients with lymphoma that did not proceed to HSCT died (n = 4). Of those proceeding to HSCT, survival was 39% (7/18) for patients with a history of lymphoma and 65% (17/26) for those without (P = .08). Both patients with vascular involvement died (cerebral aneurysm [n = 1], lymphocytic myocarditis [n = 1]). Similarly, both patients with HVLPD did not survive (cerebral aneurysm [n = 1], transplant-associated thrombotic microangiopathy [n = 1]).
At a mean of 14 months (median 9 months) of follow-up since HSCT, 47% (21/44) of patients are in complete remission with no detectable EBV viremia. Six percent (3/47) are alive with persistent host hematopoietic cells as measured by chimerism. One of these patients subsequently developed an EBV+ lymphoma and is receiving chemotherapy. Forty-five percent (20/44) of patients died after HSCT; 75% (15/20) died due to disease relapse, including multiple patients with HLH reactivation associated with rapidly progressive lymphoproliferative disease (supplemental Table 2). Three patients died during their conditioning regimen but before HSCT (1 with sepsis, 1 with a gastrointestinal bleed related to disease, and 1 with HLH reactivation). Transplant-related mortality was relatively low (10% [5/48]) for a cohort with such severe disease before HSCT; 3 deaths were due to sepsis (including the 1 before stem cell infusion) and 2 were due to thrombotic microangiopathy.
Thirty-eight percent (18/47) of patients received EBV-specific T cells after HSCT. Some patients received infusions as a prophylactic measure post-HSCT (particularly when donor derived), whereas others were given when patients relapsed. Sixty-one percent (n = 11) of these patients survived; 7 patients died. This survival rate was not different from the rest of the transplanted group. Of the 7 deaths, 5 (71%) were due to disease progression, 1 due to transplant related mortality, and 1 due to a drug overdose while in full remission.
Of the 8 patients who did not undergo HSCT, 6 died. All died of progressive CAEBV, including episodes of intracranial hemorrhage, multiorgan failure, and/or lymphocytic myocarditis. Three had EBV+ lymphoma at autopsy. Of the 2 patients who have survived without HSCT, 1 presented with HLH. She was found to have 2 million copies of EBV/mL of blood, with EBV primarily in T cells. She was treated with prednisolone and 3 courses of bortezomib/ganciclovir with a partial response; at 16 months from the onset of HLH, she has no further HLH symptoms but remains viremic (79 000 copies EBV/mL at last assessment). The second patient also presented with HLH and T-cell disease and had a complete response to 9 doses of nivolumab; 2 months since his last dose, he is asymptomatic with undetectable blood EBV PCR. These patients, presenting with T-cell disease and HLH, may be on a spectrum of disease between EBV T-cell–associated HLH that may resolve with chemotherapy,18 and CAEBV, which requires HSCT.
Discussion
We report 57 patients with CAEBV, which to our knowledge is the largest cohort to date of patients outside of Asia. Unlike other reports, one-third (22/57) of our patients were white. Historically, this has been a disease of persons with Asian and/or Central American ancestry. Our cohort suggests that CAEBV may be more common in the white population than previously described. Additionally, 12% (7/57) of patients in the cohort were Native American, a high number considering they represent less than 4% of the total North American population.19,20 Native Americans are believed to have migrated from Asia over the Bering land bridge 20 000 years ago.21 Thus, similar genetic backgrounds in Native Americans and Asians may predispose them to CAEBV. It has been postulated that disease severity may correlate with ethnicity, with CAEBV outside Asia being less severe.6 In our cohort, differences in ethnicity were not associated with differences in survival.
Most patients in our cohort had T-cell CAEBV. This differs from the Asian experience, where NK and T-cell CAEBV are equally distributed.2 There was no obvious association between ethnic group and cell-type involvement in our cohort. In Asia, NK cell CAEBV is reported to have a better prognosis than T-cell CAEBV.4 In contrast, we observed similar mortality rates regardless of NK or T-cell disease. Unlike our prior report,9 we excluded patients with B-cell CAEBV. With the use of whole exome sequencing, we found that some of the patients in the prior report with B-cell CAEBV had germline pathologic mutations in genes known to be associated with severe EBV disease, including PI3KCD, MAGT1, STXBP2, and PRF1. None of the patients in the prior report with NK or T-cell CAEBV had germline pathologic mutations.
A striking finding is that long-term survival was low in our cohort, with near 50% mortality. In studies from Asia, the survival rate is higher, ranging from 60% to as high as 90%.8,10 This may be due to the relative rarity of CAEBV in the United States and Europe, leading to late recognition, more advanced disease, and a higher mortality risk with HSCT. Our cohort is likely skewed toward those with more severe disease, as they were identified only after referral for cellular therapy or CAEBV protocols. Several patients had symptoms for months to years before interventions. In addition, over 70% of our patients were >8 years at diagnosis, which has been associated with a worse prognosis.4 This selection bias is likely an important reason for the decreased survival of the cohort. Although patients undergoing HSCT were statistically more likely to survive, mortality was still high.
Development of lymphoma showed a trend with higher risk of death in this cohort. Indeed, survival for those without lymphoma (65%) was comparable to Asian studies. Additionally, all patients with HVLPD (n = 2) and/or aneurysm secondary to disease (n = 2) died. This suggests such presentations herald a worse prognosis, though the small numbers limit our ability to provide statistical analysis.
Even in those without the above complications, most patients died due to persistent CAEBV despite multiple regimens used to control their disease and frequent use of HSCT. This indicates a critical need to develop better approaches for disease control. Although our numbers are small, medications that directly target T-cell activation may be more likely to confer disease control. In our cohort, patients that received emapalumab, nivolumab, and alemtuzumab had full responses and most were long-term survivors. Recent data suggests nivolumab is a therapeutic option in NK/T-cell–driven disorders, including lymphoma and adult HLH.22,23 Thus, PD-1 inhibition is an attractive target, along with other T-cell immunomodulatory drugs. Expert opinion has been to consider drugs that eradicate T cells, such as alemtuzumab, only before planned HSCT due to concern for the drug impairing T-cell control of EBV disease and worsening CAEBV. This was not noted in our cohort, although patients receiving alemtuzumab proceeded to HSCT shortly after starting the drug. Future studies focusing on management of disease with these interventions are important.
Rituximab was frequently used, with little response. Given rituximab’s mechanism of action, it is expected that those with NK and/or T-cell disease would not show a benefit. Because CAEBV is rare outside of Asia, and rituximab is used often in EBV-mediated B-cell disorders such as posttransplant lymphoproliferative disease, its use was likely due to an under-appreciation that EBV was predominantly in NK or T-cells. Persistently elevated EBV viremia in a nonimmunocompromised host, however, is abnormal and should prompt consideration of a diagnosis of CAEBV, particularly in those who fail to respond to rituximab.
We hypothesized that disease control before HSCT is critical for its success. In this retrospective review, defining disease control was challenging. EBV viral load was collected as an imperfect surrogate of disease activity. A reduced viral load (defined as <10 000 EBV copies/mL of blood) before HSCT did not result in a survival advantage. Control of hyperinflammation to reduce disease severity, which is essential for treating EBV-associated HLH and maintaining good organ function, is likely important before HSCT. More studies are needed to identify treatments that will best control disease in order to achieve improved long-term survival.
Three patients in our cohort are alive after HSCT but with mixed donor chimerism. Management of these cases remains a point of contention among experts. Some recommend these patients be monitored without intervention in the absence of clinical issues, whereas others recommend early consideration for further treatments, including second transplant.8 We believe high-level donor T-cell chimerism is important to sustain disease control post-HSCT, though our numbers preclude our ability to analyze this further. The poor survival in our cohort leads us to advocate for early HSCT in patients with CAEBV, as well as aggressive management of elevated EBV loads after HSCT, as morbidity and mortality are likely to be improved before the development of complications that may be irreversible or preclude a second HSCT. The high rate of death due to relapse after transplant (75%) indicates the need for better control of disease. This could be either due to poor control before HSCT or the need for a more intense preparative regimen, although myeloablative regimens did not result in improved survival. Post-transplant EBV-specific T cells may be helpful as adjunctive prophylactic treatment post-HSCT.16,24 However, survival in those receiving EBV-specific T cells did not differ from the overall survival of the cohort. EBV-specific T cells were used both as prophylaxis and for active disease in this cohort, which hinders our ability to fully answer this question. Myeloid-derived suppressor cells may be another important therapeutic target in this setting25 and are being considered for future T-cell studies in which the tumor microenvironment might be modified to improve the ability of cytotoxic T lymphocytes to kill EBV-infected cells.
A limitation of our study is the relatively small number of patients, which makes subgroup comparisons less likely to be significant. Our study, however, is the largest reported to date of patients outside of Asia and accrued patients over a decade. Larger studies will be needed to observe statistical differences among subgroups.
In conclusion, CAEBV remains a challenging disorder to treat, with high mortality rates once severe complications develop. Various approaches have been tried to control disease before HSCT; none of the currently used treatments result in consistent remission. T-cell modulating therapies may enhance disease control; future studies will need to address this question in a controlled setting. HSCT remains the only therapeutic intervention that can cure the disease, but relapses can occur. Earlier use of HSCT, better disease control, and preservation of organ function before transplant, as well as early intervention if EBV loads rise after HSCT, are likely critical to improved outcomes.
Acknowledgments
This work was supported by the intramural research program of the National Institute of Allergy and Infectious Diseases and partially by the National Cancer Institute, National Institutes of Health (5P50CA126752).
Authorship
Contribution: B.J.D.S. and C.M.B. designed the study; L.C. helped collect and summarize all data; B.J.D.S. supervised and wrote the first draft of the study; C.M.B. and J.I.C. helped with editing the manuscript. B.J.D.S., T.J., C.B., D.B., S. Chandra, S. Chandrakasan, W.C., H.L.E., A.V.G., R.H.G., R.G., D.H., S.I., K.M., H.K.M., W.O., M.P., N.C.P., C.Q., T.Q., N.R., D.S., E. Shereck, E. Stenger, and J.I.C. contributed patient data to the study; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: C.B. has pending patent applications describing the use of engineered NK cells to enhance tumor targeting and has received research funding from Merck, Sharp, and Dohme, Inc.; Bristol Myers Squibb; and Kiadis Pharma. E. Stenger has served as an elected physician member of the board of directors of the International Society for Cell and Gene Therapy and as an elected member of the scientific executive committee for the Sickle Cell Transplant Advocacy and Research Alliance, which has received research funding from bluebird bio, Inc. M.B.J. is a consultant for Atara therapeutics and Sobi. H.E.H. has equity in Allovir and Marker Therapeutics; has served on advisory boards for Tessa Therapeutics, Gilead Biosciences, Novartis, and Kiadis; and has research support from Tessa Therapeutics and Kuur Therapeutics. C.M.B. is a cofounder and on the scientific advisory boards for Catamaran Bio and Mana Therapeutics with stock and/or ownership, is on the board of directors for Caballeta Bio with stock options, has stock in Neximmune and Repertoire Immune Medicines, and serves on the Data and Safety Monitoring Boards for Sobi. The remaining authors declare no competing financial interests.
Correspondence: Blachy J. Dávila Saldaña, Children’s National Hospital, 111 Michigan Ave NW, Washington, DC 20010; e-mail: bjdavila@childrensnational.org.

