

Key Points
IPF has a typical clinical presentation with involvement of the lower limbs corresponding to vascular territories.
The antithrombin level and platelet count at diagnosis seemed to be associated with severe complications in IPF.

Abstract
Idiopathic purpura fulminans (IPF) is a rare but severe prothrombotic coagulation disorder that can occur after chickenpox or human herpesvirus 6 (HHV-6) infection. IPF leads to an autoantibody-mediated decrease in the plasma concentration of protein S. We conducted a retrospective multicenter study involving patients with IPF from 13 French pediatric centers and a systematic review of cases in published literature. Eighteen patients were included in our case series, and 34 patients were included as literature review cases. The median age was 4.9 years, and the diagnostic delay after the first signs of viral infection was 7 days. The lower limbs were involved in 49 patients (94%) with typical lesions. In all, 41 patients (78%) had a recent history of varicella-zoster virus infection, and 7 patients (14%) had been infected by HHV-6. Most of the patients received heparin (n = 51; 98%) and fresh frozen plasma transfusions (n = 41; 79%); other treatment options were immunoglobulin infusion, platelet transfusion, corticosteroid therapy, plasmapheresis, and coagulation regulator concentrate infusion. The antithrombin level and platelet count at diagnosis seemed to be associated with severe complications. Given the rarity of this disease, the creation of a prospective international registry is required to consolidate these findings.
Introduction
Idiopathic purpura fulminans (IPF) is a rare syndrome, first described by D’Angelo et al1 and Levin et al.2 Unlike acute infectious purpura fulminans, which is the result of an active and severe bacterial infection, IPF is related to an acquired protein S (PS) deficiency secondary to viral infection by varicella-zoster virus (VZV) or human herpesvirus 6 (HHV-6) that promotes the production of PS autoantibodies. The most likely mechanism is cross-reactivity against the virus and PS via molecular mimicry.3 IPF has been mainly described in case reports or small case series.2,4-6 Although IPF can be complicated by extensive skin necrosis and limb amputation, treatment for IPF is not well defined.
Our study sought to describe patient characteristics, treatments, and outcomes of IPF by pooling the results of a French multicenter case series along with characteristics of patients described in the literature. Our secondary objective was aimed at identifying prognostic factors. Here, we propose a decision tree for diagnostic management and leads for treating IPF.
Methods
Case series
This was a multicenter study at 13 French pediatric academic centers (Montpellier, Marseille, Reims, Grenoble, Poitiers, Paris-Necker, Paris-Robert-Debré, Nantes, Nice, Nancy, Tours, Strasbourg, and Lyon). The main French centers of intensive pediatric hematology were contacted to participate in this retrospective study. The inclusion criteria comprised the presence of clinical IPF with a low level of PS and/or a history of viral infection in the 15 days before IPF in children younger than age 15 years. The exclusion criteria comprised previously published cases, other causes of infectious purpura fulminans (eg, pneumococcus, meningococcus), severe congenital protein C (PC) and PS deficiency, cancer, and hematologic malignancies. The data were collected via a survey and from information in the patients’ medical records. Data were collected on the clinical and the biological characteristics of patients at admission and their treatments and outcomes. The time to diagnosis was calculated from the first signs of viral infection to the diagnosis of IPF. The extent of lesions was evaluated from clinical descriptions and photographs. The PS level is provided according to which of the 3 assay methods was used (activity, free antigen, or total antigen) and is expressed as a percentage. Antithrombin (AT) and PC are expressed as the percentage of normal activity, the fibrinogen level is expressed in grams per liter, and the platelet count is provided in grams per liter when available on admission. D-dimer levels were considered to be increased if they exceeded the laboratory standard. The diagnosis of viral infection was clinical for chickenpox with or without biological confirmation of VZV, and it was biological by serology or polymerase chain reaction for HHV-6.
The study was approved by the institutional review board of Montpellier University Hospital on behalf of all participating centers and was conducted in accordance with the provisions of the Declaration of Helsinki.
Literature review cases
In the absence of a case series with sufficient clinical and biological data on this pathology, we decided to pool clinical cases from the literature despite the limitations of this methodology. Our results consider the limitation that the previously published cases may not represent all cases at those individual institutions and may not be fully representative of the study population. The MEDLINE (1946 to July 1, 2019) database was used with the following keywords: “idiopathic purpura fulminans,” “acquired protein S deficiency,” and “anti-protein S antibodies.” Our search was limited to children. All articles that met the inclusion criteria and that presented sufficient data were included. The same data were collected as would be collected in a retrospective case series.
Statistical analysis
The continuous variables are presented as medians with the minimum and maximum values, and the dichotomous variables are relative to the total number. For each data set, we show the number of missing data when appropriate.
Incidences of extensive necrosis leading to distal amputation and skin necrosis were classified as severe thrombotic complications. To determine factors associated with the occurrence of severe complications, we reported the distribution of the median of each factor according to the patients’ outcomes (severe thrombotic complications). The median distributions were compared using Student t test if they had a normal distribution; otherwise, we used Mann-Whitney U tests. We used multiple univariable logistic regression models with the sociodemographic and the biological factors at the time of diagnosis. The regression models for the biological factors are inverted to present the effect when they decreased. We then constructed a multivariable logistic regression model using a stepwise selection of variables with a P value <.2 in the univariable analyses as well as the patient’s age.
Results
Data extraction
Our case series, which included data from January 1, 1989, to December 31, 2018, included 18 children from 13 French pediatric units (Montpellier, n = 6; Marseille, Reims, Grenoble, Poitiers, Paris-Necker, Paris-Robert-Debré, Nantes, Nice, Nancy, Tours, Strasbourg, and Lyon, n = 1). In the literature review, we found 27 relevant articles published between 1992 and 2019. We excluded 2 of these articles because they had insufficient data. In the remaining 25 articles, we excluded 4 cases from 2 case series, 1 for lack of data and 3 that did not meet the inclusion criteria. Ultimately, we retained 34 cases in 25 articles as literature review cases.1-3,5-26 The data presented below are the pooled results of 52 cases from both the case series and the literature review, unless otherwise indicated. The case series results and the literature review results are presented separately in Table 1.
Patient characteristics, clinical presentation, type of virus, biology at diagnosis, treatment, and outcomes
| Case series (n = 18) | Literature review cases (n = 34) | Pooled data (n = 52) | Missing data | ||||
|---|---|---|---|---|---|---|---|
| No. (%) | Median (range) | No. (%) | Median (range) | No. (%) | Median (range) | No. (%) | |
| Patient characteristics | |||||||
| Age, y | 4.4 (1.5-6) | 5.2 (1.8-11) | 4.9 (1.5-11) | — | |||
| Male sex | 7 (39) | 20 (59) | 27 (52) | — | |||
| Clinical presentation | |||||||
| Time to diagnosis, d | 6 (2-17) | 8 (5-15) | 7 (2-17) | — | |||
| Lower limb | 16 (89) | 33 (97) | 49 (94) | — | |||
| Calves only | 3 (17) | 10 (29) | 13 (25) | — | |||
| Extended leg | 13 (72) | 23 (68) | 36 (69) | — | |||
| Upper limb | 2 (11) | 5 (14) | 7 (13) | — | |||
| Genitalia | 1 (5.6) | 3 (9) | 4 (8) | — | |||
| Torso | 3 (17) | 8 (23) | 11 (21) | — | |||
| Virus | |||||||
| HHV-6 | 3 (17) | 1 (3) | 4 (8) | — | |||
| VZV | 11 (61) | 30 (86) | 41 (78) | — | |||
| Unknown | 4 (22) | 4 (11) | 7 (14) | — | |||
| Biology at diagnosis | |||||||
| PS activity, % | 5 (1-28) | 2.5 (1-25) | 4 (1-28) | 24 (52) | |||
| PS free antigen, % | 6 (1-16) | 1 (1-9) | 1 (1-16) | 24 (46) | |||
| PS total antigen, % | 4.5 (1-32) | 5.5 (1-62) | 5 (1-62) | 22 (42) | |||
| Anti-PS antibody | 13 (72) | 17 (50) | 30 (58) | — | |||
| PC activity, % | 43 (14-131) | 54 (14-100) | 49.5 (14-131) | 4 (8) | |||
| Antithrombin level, % | 85 (45-130) | 86 (48-115) | 85 (45-130) | 11 (21) | |||
| Fibrinogen level, g/L | 1.85 (0.35-3.4) | 0.5 (0.1-2.38) | 0.88 (0.1-3.4) | 10 (19) | |||
| Platelet count, ×109/L | 65 (10-250) | 166 (14-302) | 150 (10-302) | 8 (15) | |||
| Increase in D-dimer | 14 (78) | 16/18 (89) | 30/36 (83) | 17 (33) | |||
| Treatment | |||||||
| Heparin | 18 (100) | 33 (97) | 51 (98) | — | |||
| Corticosteroid | 3 (17) | 13 (37) | 16 (31) | — | |||
| Fresh frozen plasma | 13 (72) | 29 (83) | 41 (79) | — | |||
| Polyvalent Ig | 8 (44) | 11 (31) | 20 (38) | — | |||
| Plasmapheresis | 7 (39) | 6 (17) | 13 (25) | — | |||
| Coagulation regulator* | 5 (28) | 4 (11) | 12 (23) | — | |||
| Outcomes | |||||||
| Amputation | 6 (33) | 8 (24) | 14 (27) | — | |||
| Skin necrosis with graft | 5 (28) | 10 (29) | 15 (29) | — | |||
| Veinous thromboembolism | 6 (33) | 11 (32) | 17 (33) | — | |||
| Hemorrhagic complication | 2 (11) | ND | ND | — | |||
| Case series (n = 18) | Literature review cases (n = 34) | Pooled data (n = 52) | Missing data | ||||
|---|---|---|---|---|---|---|---|
| No. (%) | Median (range) | No. (%) | Median (range) | No. (%) | Median (range) | No. (%) | |
| Patient characteristics | |||||||
| Age, y | 4.4 (1.5-6) | 5.2 (1.8-11) | 4.9 (1.5-11) | — | |||
| Male sex | 7 (39) | 20 (59) | 27 (52) | — | |||
| Clinical presentation | |||||||
| Time to diagnosis, d | 6 (2-17) | 8 (5-15) | 7 (2-17) | — | |||
| Lower limb | 16 (89) | 33 (97) | 49 (94) | — | |||
| Calves only | 3 (17) | 10 (29) | 13 (25) | — | |||
| Extended leg | 13 (72) | 23 (68) | 36 (69) | — | |||
| Upper limb | 2 (11) | 5 (14) | 7 (13) | — | |||
| Genitalia | 1 (5.6) | 3 (9) | 4 (8) | — | |||
| Torso | 3 (17) | 8 (23) | 11 (21) | — | |||
| Virus | |||||||
| HHV-6 | 3 (17) | 1 (3) | 4 (8) | — | |||
| VZV | 11 (61) | 30 (86) | 41 (78) | — | |||
| Unknown | 4 (22) | 4 (11) | 7 (14) | — | |||
| Biology at diagnosis | |||||||
| PS activity, % | 5 (1-28) | 2.5 (1-25) | 4 (1-28) | 24 (52) | |||
| PS free antigen, % | 6 (1-16) | 1 (1-9) | 1 (1-16) | 24 (46) | |||
| PS total antigen, % | 4.5 (1-32) | 5.5 (1-62) | 5 (1-62) | 22 (42) | |||
| Anti-PS antibody | 13 (72) | 17 (50) | 30 (58) | — | |||
| PC activity, % | 43 (14-131) | 54 (14-100) | 49.5 (14-131) | 4 (8) | |||
| Antithrombin level, % | 85 (45-130) | 86 (48-115) | 85 (45-130) | 11 (21) | |||
| Fibrinogen level, g/L | 1.85 (0.35-3.4) | 0.5 (0.1-2.38) | 0.88 (0.1-3.4) | 10 (19) | |||
| Platelet count, ×109/L | 65 (10-250) | 166 (14-302) | 150 (10-302) | 8 (15) | |||
| Increase in D-dimer | 14 (78) | 16/18 (89) | 30/36 (83) | 17 (33) | |||
| Treatment | |||||||
| Heparin | 18 (100) | 33 (97) | 51 (98) | — | |||
| Corticosteroid | 3 (17) | 13 (37) | 16 (31) | — | |||
| Fresh frozen plasma | 13 (72) | 29 (83) | 41 (79) | — | |||
| Polyvalent Ig | 8 (44) | 11 (31) | 20 (38) | — | |||
| Plasmapheresis | 7 (39) | 6 (17) | 13 (25) | — | |||
| Coagulation regulator* | 5 (28) | 4 (11) | 12 (23) | — | |||
| Outcomes | |||||||
| Amputation | 6 (33) | 8 (24) | 14 (27) | — | |||
| Skin necrosis with graft | 5 (28) | 10 (29) | 15 (29) | — | |||
| Veinous thromboembolism | 6 (33) | 11 (32) | 17 (33) | — | |||
| Hemorrhagic complication | 2 (11) | ND | ND | — | |||
ND, not diagnosed.
Antithrombin and activated PC.
Characteristics at admission
The median age at diagnosis was 4.9 years (range, 1.5-11 years) and the ratio of males:females was 0.52. The time to diagnosis was 7 days (range, 2-17 days). The clinical presentation was an ecchymotic or necrotic purpura of a lower limb (n = 49; 94%), the calves only (n = 13; 25%), an upper limb (n = 7; 13%), the torso (n = 11; 21%), or the genitalia (n = 4; 8%). Several patients presented with identical vascular territories such as “sock-like” lesions of the calves and/or necrosis of the anterolateral side of the thigh (Figure 1).
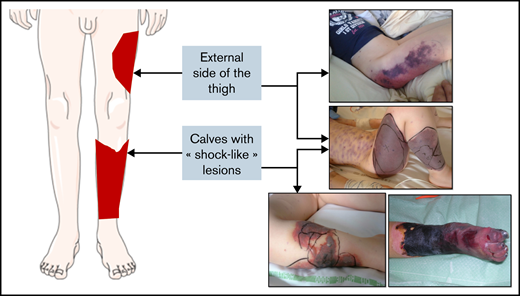
Schematic depiction of typical lower limb lesions observed in many cases, with corresponding photographs of affected areas.
Schematic depiction of typical lower limb lesions observed in many cases, with corresponding photographs of affected areas.
A diagnosis of viral infection was confirmed for 45 patients (86%): chickenpox (n = 41; 78%) or HHV-6 (n = 4; 8%). PS deficiency was confirmed in all of the patients. The median PS activity level was 4% (range, 1%-28%). The median free PS antigen level was 1% (range, 1%-16%), and the median total PS antigen level was 5% (range, 1%-62%). The presence of anti-PS antibodies was confirmed in 30 patients (58%). At admission, the median AT activity level was 85% (range, 45%-130%), the median PC activity level was 49.5% (range, 14%-131%), the median fibrinogen level was 0.88 g/L (range, 0.1-3.4 g/L), and the median platelet count was 150 × 109/L (range, 10-302 g/L). D-dimer levels were tested in 36 (69%) of 52 cases, and they increased in 30 (83%) of 36 cases.
Treatment and outcomes
Treatments for IPF included intravenous (IV) heparin (n = 51; 98%), IV fresh frozen plasma (FFP) (n = 41; 79%), IV polyvalent immunoglobulins (Ig’s) (n = 20; 38%), IV corticosteroids (n = 16; 31%), plasmapheresis (n = 13; 25%), and IV coagulation inhibitor concentrates (AT or activated PC) (n = 12; 23%).
The PS levels normalized in all the patients, albeit with variable time frames. The PS levels started to increase at 10 days (range, 2-49 days), with a normalization median delay of 60 days (range, 6-120 days). There were no deaths, although 25 patients (47%) had serious thrombotic complications, including distal amputation (n = 14; 27%) or skin necrosis with grafting (n = 15; 29%) (4 patients required both); 17 patients (33%) developed venous thromboembolism, and 2 patients had a hemorrhage (1 subarachnoid and 1 pulmonary alveolar).
We analyzed the relationship between several factors and the occurrence of severe complications in the overall cohort. At the time of diagnosis, the median AT activity level (79% vs 101%; P < .001) and the median platelet count (51.5 × 109/L vs 188.5 × 109/L; P < .001) were significantly lower in patients with severe complications. Univariable analyses confirmed the results with odds ratios (ORs) of 1.08 (95% confidence interval [CI], 1.03-1.14; P = .003) for the median AT activity level and 1.02 (95% CI, 1.01-1.02; P = .001) for the median platelet count. In multivariable analysis, a correlation was found between severe complications and the median AT activity level, with an OR of 1.07 (95% CI, 1.01-1.14; P = .03) and the median platelet count with an OR of 1.01 (95% CI, 1.00-1.02; P = .04). All results are presented in Figure 2 and Table 2.
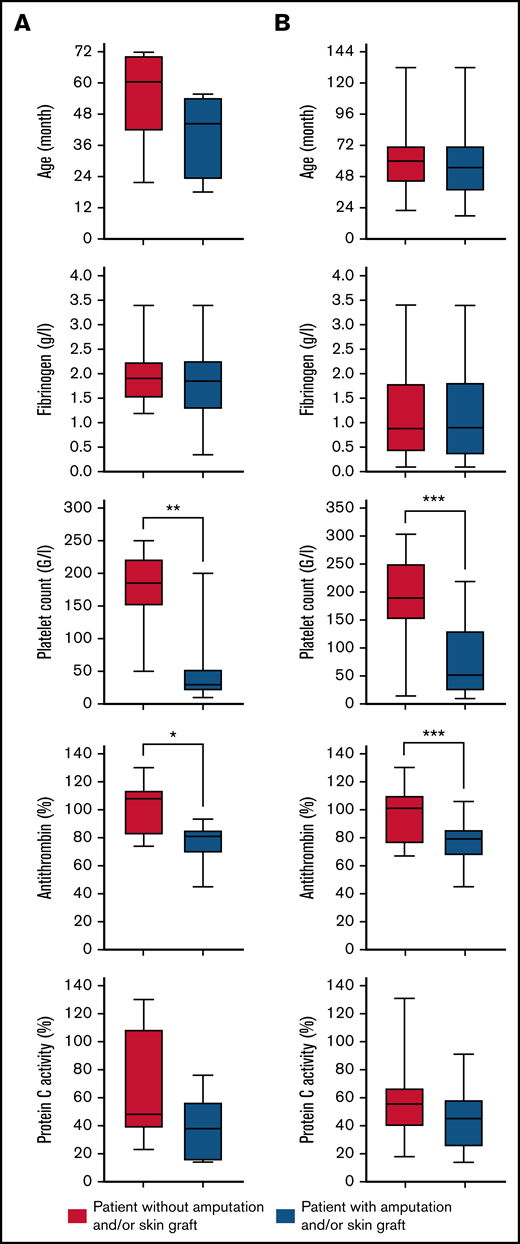
Box plot showing the age distribution, fibrinogen level, platelet count, antithrombin level, and PC activity at the time of diagnosis according to patient outcome (with or without amputation and/or skin graft). The box represents the interquartile (25th to 75th percentiles) range. The horizontal line in the box represents the median. The whiskers represent the maximum and minimum values (A) for case series patients and (B) for all patients (cases series and literatures review cases). The P value was estimated using Mann-Whitney nonparametric U test or Student t test. *P < .05; **P < .01; **P < .001.
Box plot showing the age distribution, fibrinogen level, platelet count, antithrombin level, and PC activity at the time of diagnosis according to patient outcome (with or without amputation and/or skin graft). The box represents the interquartile (25th to 75th percentiles) range. The horizontal line in the box represents the median. The whiskers represent the maximum and minimum values (A) for case series patients and (B) for all patients (cases series and literatures review cases). The P value was estimated using Mann-Whitney nonparametric U test or Student t test. *P < .05; **P < .01; **P < .001.
Results of univariable and multivariable logistic regression for the risk of severe thrombotic complications (amputation, skin necrosis with grafting)
| OR | 95% CI | P | Adjusted OR | 95% CI | P | |
|---|---|---|---|---|---|---|
| Female sex | 0.60 | 0.12-3.00 | .53 | — | — | — |
| Age, y* | ||||||
| <4.5 | 2.02 | 0.54-7.49 | .49 | — | — | — |
| 4.5-6 | 1.83 | 0.43-7.77 | .69 | — | — | — |
| >6 | Ref | Ref | Ref | — | — | — |
| Antithrombin† | 1.08 | 1.03-1.14 | .003 | 1.07 | 1.01-1.14 | .03 |
| Fibrinogen† | 1.08 | 0.55-2.13 | .82 | — | — | — |
| Platelets† | 1.02 | 1.01-1.02 | .001 | 1.01 | 1.00-1.02 | .04 |
| PC† | 1.02 | 1.00-1.05 | .08 | — | — | — |
| OR | 95% CI | P | Adjusted OR | 95% CI | P | |
|---|---|---|---|---|---|---|
| Female sex | 0.60 | 0.12-3.00 | .53 | — | — | — |
| Age, y* | ||||||
| <4.5 | 2.02 | 0.54-7.49 | .49 | — | — | — |
| 4.5-6 | 1.83 | 0.43-7.77 | .69 | — | — | — |
| >6 | Ref | Ref | Ref | — | — | — |
| Antithrombin† | 1.08 | 1.03-1.14 | .003 | 1.07 | 1.01-1.14 | .03 |
| Fibrinogen† | 1.08 | 0.55-2.13 | .82 | — | — | — |
| Platelets† | 1.02 | 1.01-1.02 | .001 | 1.01 | 1.00-1.02 | .04 |
| PC† | 1.02 | 1.00-1.05 | .08 | — | — | — |
Ref., reference.
Age did not have a normal distribution in our series; we considered 3 age brackets according to the distribution of the patients.
Regression models for biological factors are inverted to present the effect when they decreased.
Decision tree for diagnosis and management of disease
We propose simple diagnostic criteria to help with diagnosis of IPF (summarized in Figure 3).
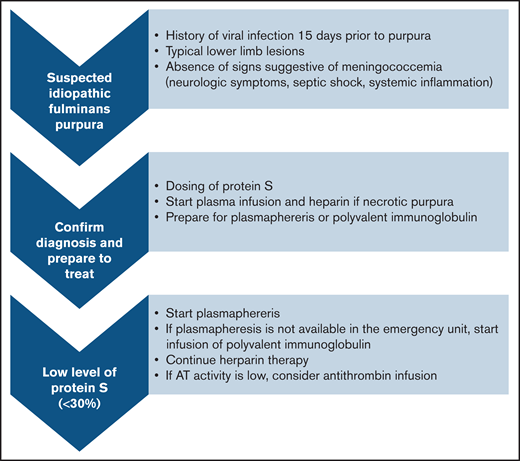
Proposed algorithm for diagnostic and treatment of idiopathic fulminans purpura.
Proposed algorithm for diagnostic and treatment of idiopathic fulminans purpura.
When to suspect IPF.
IPF is a serious condition that requires urgent management. The existence of a VZV or HHV-6 infection within 15 days before the purpura are among the most relevant disease history data. On clinical examination, most patients (94%) had lower limb involvement with typical lesions. Because IPF is very rare, it is necessary to eliminate the most frequent causes of purpura fulminans such as bacterial infections, which tend to present as states of severe sepsis or even septic shock with elevation of inflammatory markers such as C-reactive protein and procalcitonin, and they can be accompanied by meningeal irritation.
How to confirm the diagnosis and start treatment quickly.
The essential criteria for distinguishing purpura fulminans from other forms of purpura and coagulopathy is the presence of a PS deficiency. Once the coagulation test has been performed, the most widely used treatments are fresh plasma infusion and heparin therapy. Because the main risk is thrombotic complication, therapy can be started immediately in the absence of hemorrhagic signs.
How to treat once the diagnosis is confirmed.
Plasmapheresis and Ig infusions seem to be the 2 most effective treatments against the PS inhibitor. It seems relevant, therefore, to administer them as soon as possible. The continuation of an anticoagulant treatment remains essential, and the addition of antithrombin concentrate can be considered in case of deficiency.
Discussion
IPF is a rare disease that affects young children, with potentially severe sequelae such as distal amputations and extensive skin necrosis. Our case series confirm the natural history described in the literature. IPF starts 7 days after a VZV or HHV-6 infection and leads to thrombotic complications that mainly affect the lower limbs. Anti-PS autoantibodies were found in more than half the patients, thus suggesting the autoimmune origin of the disease linked with an infectious antigenic stimulus.2 In our series, as in the literature, the autoantibodies seem to fully disappear within a few weeks, irrespective of the treatment used. Two studies have reported an asymptomatic decrease in the level of PS in the context of infection by VZV, but there is no association with antibodies or thrombotic symptoms.27,28 The prevalence of anti-PS antibodies is not known nor are the factors that can favor progression to this acute and severe form of thrombophilia, because chickenpox and HHV-6 infections are extremely common and IPF is the exception.
The clinical lesions revealed a degree of similarity among patients because the anatomical locations of the necrosis seemed to overlap with the anatomical vascular territories, such as perforating branches of the fibular artery, the tibial anterior artery, and the deep femoral artery. This suggests a complete thrombotic obstruction of the corresponding perforasome without replacement by adjacent areas.29-32 These anatomical data are consistent with the presence of targeted thrombosis in the distal limb circulation.
The biology at diagnosis reflected either a normal condition or signs of consumption (lowering of fibrinogen and platelets, elevation of D-dimers), which may be compatible with a diagnosis of disseminated intravascular coagulation (DIC).33 A severe decrease in the level of PS in most patients allows differentiation from conventional DIC. PS assays should, therefore, be performed promptly in case of any suspicion of IPF.
Most of the clinical cases described a rapid progression to severe thrombotic complications. Hence, IPF treatment must be effective to prevent the occurrence of thrombosis and to eliminate the antibodies. In the literature, heparin anticoagulation and FFP infusion are the most used treatments, whereas the effectiveness of Ig infusions and plasmapheresis to decrease the inhibitor has been suggested in several case reports.2,12,20 In our study, the AT level and platelet count at diagnosis seemed to be associated with severe complications. During conventional DIC, the AT level decreases by consumption at an early phase of the DIC, and it can predict the outcome.34 It raises the issue of AT concentrate infusions in patients with decreased AT activity levels at diagnosis, as proposed for DIC.35
To date, there are no targeted recommendations for the diagnostic and therapeutic management of IPF in the context of PS antibodies. We propose straightforward criteria for diagnosing and managing these patients in a fast and effective way to limit the sequelae related to severe thrombotic complications (Figure 3). The key points of diagnosis are the clinical history, with the occurrence of a recent varicella infection or elements in favor of a primary HHV-6 infection, and the presence of purpura lesions of the lower limbs with involvement in the vascular territory. The absence of signs of shock, more commonly found in DIC, is also an important factor, because signs of septic shock and systemic inflammation (elevated C-reactive protein and procalcitonin) are observed in acute infectious purpura fulminans. Conventional coagulation tests do not allow a clear-cut decision to be made, and only an emergency determination of PS allows confirmation of the diagnosis. Detection of an antibody takes too much time; determination of PS deficiency is sufficient for making an emergency diagnosis. As soon as the diagnosis is suspected or confirmed, treatment with heparin anticoagulation and FFP infusion should be started without delay. Plasmapheresis and Ig infusions can be administered in a timely manner.
A major limitation of our study is the small number of patients. The literature review cases make it possible to increase the number of patients, but that adds a selection bias because only the most significant cases are published. The similarity of the results between our retrospective case series and the literature review cases is reassuring in this regard.
Given the rarity of this disease, a prospective international registry might be an appropriate solution to progress the understanding and management of IPFs.
Authorship
Contribution: A.T. helped design the research, collected and analyzed data, and helped write the paper; O.D. helped design the research and collected data; E.B. helped design the research; A.Z. and M.R. collected data; J.B., J.-F.S., T.K., and M.D. helped write the paper; L.M. analyzed the data; E.J. and C.B.-A. helped designed the research, analyzed the data, and helped write the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Alexandre Theron, Montpellier University Hospital, 371 Av. du Doyen Gaston Giraud, 34090 Montpellier, France; e-mail: a-theron@chu-montpellier.fr.

