

Key Points
HRPII binds vascular GAGs and activates coagulation and inflammation.
AT exerts protective effects against HRPII in cellular and animal models.

Abstract
Plasmodium falciparum-derived histidine-rich protein II (HRPII) has been shown to inhibit heparin-dependent anticoagulant activity of antithrombin (AT) and induce inflammation in vitro and in vivo. In a recent study, we showed that HRPII interacts with the AT-binding vascular glycosaminoglycans (GAGs) not only to disrupt the barrier-permeability function of endothelial cells but also to inhibit the antiinflammatory signaling function of AT. Here we investigated the mechanisms of the proinflammatory function of HRPII and the protective activity of AT in cellular and animal models. We found that AT competitively inhibits the GAG-dependent HRPII-mediated activation of NF-κB and expression of intercellular cell adhesion molecule 1 (ICAM1) in endothelial cells. Furthermore, AT inhibits HRPII-mediated histone H3 citrullination and neutrophil extracellular trap (NET) formation in HL60 cells and freshly isolated human neutrophils. In vivo, HRPII induced Mac1 expression on blood neutrophils, MPO release in plasma, neutrophil infiltration, and histone H3 citrullination in the lung tissues. HRPII also induced endothelial cell activation as measured by increased ICAM1 expression and elevated vascular permeability in the lungs. AT effectively inhibited HRPII-mediated neutrophil infiltration, NET formation, and endothelial cell activation in vivo. AT also inhibited HRPII-meditated deposition of platelets and fibrin(ogen) in the lungs and circulating level of von Willebrand factor in the plasma. We conclude that AT exerts protective effects against pathogenic effects of P falciparum-derived HRPII in both cellular and animal models.
Introduction
Malaria is a serious infectious disease that afflicts nearly 250 million people annually, with ∼1 million fatalities.1-3 Among five different types of Plasmodium species causing malaria, infection by Plasmodium falciparum is the deadliest, leading to severe malaria that is responsible for most malaria-related fatalities. Infection by P falciparum can lead to numerous serious complications, including thrombocytopenia,4 coagulopathy,5 cerebrovascular stroke,6 severe anemia,7 acute renal failure,8 and acute respiratory distress syndrome.9 Pathophysiology of severe malaria is multifactorial and remains obscure; however, increasing evidence suggests that P falciparum-mediated endothelial cell–leukocyte interaction induces vascular dysfunction, leading to activation of coagulation and inflammation.1,2,8 The course of infection begins by the parasite entering the circulation, invading host red blood cells, and undergoing an asexual growth cycle during which the parasite expresses numerous membrane and secretory proteins as an essential process for maintaining viability and virulence.1 A well-studied P falciparum-derived membrane protein is called P falciparum erythrocyte membrane protein 1, which has been demonstrated to downregulate the protein C anticoagulant pathway by binding to endothelial protein C receptor, thereby downregulating activation of protein C and its anticoagulant and antiinflammatory functions.10,11 A P falciparum-derived secretory protein that has been shown to also promote activation of proinflammatory and procoagulant pathways is histidine-rich protein II (HRPII).1,12,13 Studies have demonstrated that HRPII binds anticoagulant glycosaminoglycans (GAGs), thereby promoting coagulation pathway by inhibiting the heparan sulfate–dependent anticoagulant function of antithrombin (AT).14 Moreover, studies have found that HRPII, through activation of inflammasome, compromises integrity of tight junctions and endothelial cell permeability function, suggesting it may play a key role in pathogenesis of cerebral malaria by disrupting the blood–brain barrier.13
In a recent study, we demonstrated that HRPII inhibits antiinflammatory function of AT by competitively binding to specific AT-binding 3-O-sulfate (3-OS) containing GAGs that are involved in transmitting d-helix–dependent protective signaling function of the serpin.15 Through interaction with vascular GAGs, AT initiates antiinflammatory signaling responses by inducing prostacyclin synthesis,16 and HRPII effectively blocks this protective function of AT in endothelial cells. HRPII also inhibited barrier protective function of AT by inducing Src-dependent phosphorylation of VE-cadherin, thereby leading to disruption of adherens junction in endothelial cells.15 Here, we investigated molecular mechanisms through which HRPII promotes inflammation and coagulation in cellular and animal models and evaluated the mechanism of protective effects of AT in these systems. Results indicate the GAG-dependent binding of HRPII to endothelial cells and leukocytes induces NF-κB activation, culminating in cell surface expression of cell adhesion molecules. Furthermore, HRPII induces neutrophil extracellular trap (NET) formation in vitro that is inhibited by AT. In an in vivo system, HRPII increased Mac1 expression on blood neutrophils, enhanced their infiltration to lung tissues, and promoted NET formation. HRPII also promoted activation of coagulation and increased platelet and fibrin(ogen) deposition in lungs and von Willebrand factor (VWF) secretion in the plasma. AT and an AT mutant with higher affinity for GAGs,17 but not a signaling-defective AT mutant,18 downregulated activation of inflammation and coagulation by HRPII. These results suggest that AT may have therapeutic value against pathogenic effects of HRPII in patients infected with P falciparum.
Materials and methods
Reagents
Human plasma-derived AT was purchased from Enzyme Research Laboratories (South Bend, IN). Recombinant AT derivatives AT-N135Q and AT-4Mut were prepared as described.17-19 Recombinant HRPII was expressed and purified to homogeneity as described.15 The complete list of reagents is presented in supplementary Materials.
Neutrophil isolation and extracellular trap analysis
Blood was collected from healthy adult volunteers (following an approved institutional review board protocol) in acid-citrate-dextrose buffer. Blood neutrophils were isolated according to the Polymorphprep (Cosmo Bio, Carlsbad, CA) protocol. Isolated neutrophils were stimulated with HRPII (100 nM) in the absence or presence of AT (2.5 µM) or its variants. After 4 hours of incubation, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% TritonX-100/ phosphate-buffered saline (PBS), and processed for staining. Percent of NET forming cells was calculated based on total number of cells per field as described.20
Flow cytometry
Endothelial cells were treated with HRPII in the presence and absence of AT. Surface expression of ICAM1 was detected by flow cytometry using fluorescein isothiocyanate (FITC)-conjugated anti-ICAM1 antibody as described.21 Surface expression of Mac1 on LY6G-positive mouse blood neutrophils was determined as described.22
Histological examination and immunofluorescence
C57BL/6J mice (8-12 weeks old) were injected intraperitoneally (IP) with either saline or HRPII (10 µg/g bodyweight) with or without prior injection of AT (500 µg/mouse, 20 µg/g bodyweight). Mice were euthanized and perfused with PBS, and organs were collected for histopathological analysis.
Vascular permeability in the lung and kidney
Mice were injected with saline or HRPII in the presence or absence of AT. After 3 hours, 150 µL of 1% Evans blue dye in normal saline was injected IV. After 30 minutes, mice were euthanized and perfused with PBS, organs were collected, and vascular permeability in the lung and kidney was determined as described.23
Details of all methods, including western blotting for monitoring expression of signaling molecules and enzyme-linked immunosorbent assay (ELISA) for determining myeloperoxidase (MPO), thrombin-AT (TAT) complex, and VWF levels, are described in supplementary Materials.
Statistical analysis
Data are presented as mean ± standard error of mean (SEM) from ≥3 independent experiments. Data were analyzed by the Student t test, and group data were analyzed using analysis of variance followed by Bonferroni post hoc test using Graph Pad Prism7 (Graph Pad Prism, San Diego, CA). A P value of < .05 was considered statistically significant.
Results
AT inhibits GAG-dependent HRPII-mediated NF-κB activation in endothelial cells
HRPII has been shown to inhibit the antiinflammatory function of AT by competitive inhibition of AT binding to endothelial cell GAGs.15 In this study, we explored mechanisms of the proinflammatory signaling function of HRPII and the protective effect of AT using endothelial cells (hTERT-HUVECs). Western blot analysis of lysates of endothelial cells, treated simultaneously with both HRPII and AT, showed that HRPII increases phosphorylation of NF-κB p65 and this effect was counteracted by AT (Figure 1A). Immunofluorescence data supported these findings and showed that AT inhibits HRPII-mediated nuclear translocation of NF-κB p65 (Figure 1B). Inhibitory effect of AT was mediated through d-helix–dependent interaction of AT with cellular GAGs because the AT d-helix mutant (AT-4Mut), which has normal protease inhibitory function but is incapable of binding GAGs,18,19 exhibited no protective activity against HRPII (Figure 1B). In agreement with these results, flow cytometry data using FITC-labeled ICAM1 indicated that AT, but not AT-4Mut, inhibits HRPII-mediated upregulation of ICAM1 expression on endothelial cells (Figure 1C). The proinflammatory function of HRPII was mediated through interaction with cellular GAGs as evidenced by the GAG antagonist, surfen, blocking binding of FITC-labeled HRPII to endothelial cells (Figure 1D). Immunofluorescence data also showed that surfen blocks HRPII-mediated nuclear translocation of NF-κB p65 (Figure 1E). We previously demonstrated HRPII has a potent barrier-disruptive effect in endothelial cells.15 Surfen also inhibited the barrier-disruptive effect of HRPII (Figure 1F). We have demonstrated that surfen can also inhibit the cytoprotective activity of AT in an in vivo model.17 In agreement with HRPII and AT interacting with overlapping binding sites on GAGs, AT-N135Q,17 which, similar to β-isoform of AT, binds GAGs with 5- to 10-fold higher affinity,24,25 exhibited significantly better competitive effect in blocking proinflammatory function of HRPII in cell permeability assay. This conclusion is based on the analysis of protective effects of increasing concentrations of AT-WT and AT-N135Q on barrier-disruptive function of HRPII in endothelial cells (Figure 1G). Similar results were obtained in human pulmonary microvascular endothelial cells. Results showed both AT and surfen inhibit HRPII-mediated NF-κB p65 nuclear translocation and permeability in human pulmonary microvascular endothelial cells (supplemental Figure 1A-B).
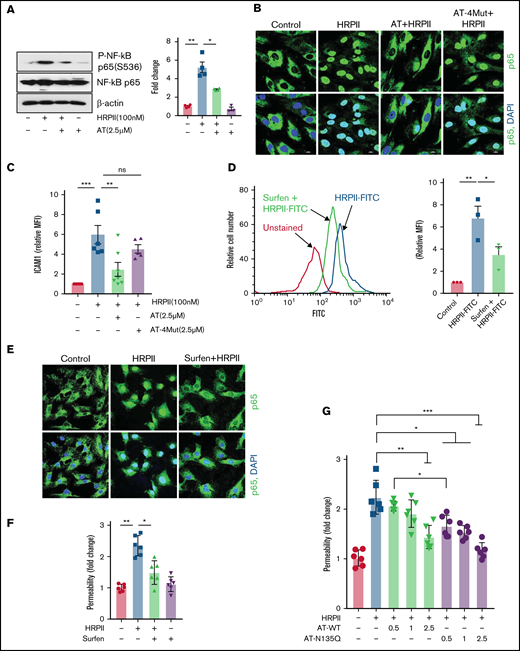
AT inhibits GAG-dependent HRPII-mediated inflammatory responses in endothelial cells. (A) Confluent endothelial cells (hTERT-HUVECs) were simultaneously treated with HRPII (100 nM) and AT (2.5 µM) for 15 min followed by analysis of phosphorylation of NF-κB p65 by western blotting. Quantitative analysis is presented. (B) Cells were treated similarly with HRPII (100 nM) and AT or AT-4Mut (2.5 µM) for 1 hour and then fixed, permeabilized, and stained with anti–NF-κB p65 antibody (rabbit) followed by Alexa Fluor 488-conjugated anti-rabbit immunoglobulin G (IgG). DAPI was used to stain the nucleus. (C) Cells were treated with HRPII and AT derivatives as described above for 4 hours followed by analysis of cell surface expression of ICAM-1 by flow cytometry. Quantitative analysis is presented. (D) FITC-conjugated HRPII (HRPII-FITC, 1 µM) binding to endothelial cells were analyzed by flow cytometry in the absence and presence of the GAG antagonist, surfen (10 µM). Quantitative analysis is presented. (E) Cells were treated with HRPII (100 nM) for 1 hour in the absence and presence of surfen (10 µM). Cells were then fixed, permeabilized, and stained with anti–NF-κB p65 (rabbit) antibody and Alexa Fluor 488-conjugated anti-rabbit IgG. DAPI was used to stain the nucleus. (F) Confluent cells were treated with HRPII in absence or presence of surfen (10 µM) followed by monitoring HRPII-mediated barrier-disruptive function through measuring the influx of albumin-bound Evans blue dye across the cell monolayer as described in "Materials and methods." (G) The same as (F) except that the competitive effect of increasing concentration of wild-type AT and AT-N135Q were monitored in presence of fixed concentration of HRPII (40 nM).
AT inhibits GAG-dependent HRPII-mediated inflammatory responses in endothelial cells. (A) Confluent endothelial cells (hTERT-HUVECs) were simultaneously treated with HRPII (100 nM) and AT (2.5 µM) for 15 min followed by analysis of phosphorylation of NF-κB p65 by western blotting. Quantitative analysis is presented. (B) Cells were treated similarly with HRPII (100 nM) and AT or AT-4Mut (2.5 µM) for 1 hour and then fixed, permeabilized, and stained with anti–NF-κB p65 antibody (rabbit) followed by Alexa Fluor 488-conjugated anti-rabbit immunoglobulin G (IgG). DAPI was used to stain the nucleus. (C) Cells were treated with HRPII and AT derivatives as described above for 4 hours followed by analysis of cell surface expression of ICAM-1 by flow cytometry. Quantitative analysis is presented. (D) FITC-conjugated HRPII (HRPII-FITC, 1 µM) binding to endothelial cells were analyzed by flow cytometry in the absence and presence of the GAG antagonist, surfen (10 µM). Quantitative analysis is presented. (E) Cells were treated with HRPII (100 nM) for 1 hour in the absence and presence of surfen (10 µM). Cells were then fixed, permeabilized, and stained with anti–NF-κB p65 (rabbit) antibody and Alexa Fluor 488-conjugated anti-rabbit IgG. DAPI was used to stain the nucleus. (F) Confluent cells were treated with HRPII in absence or presence of surfen (10 µM) followed by monitoring HRPII-mediated barrier-disruptive function through measuring the influx of albumin-bound Evans blue dye across the cell monolayer as described in "Materials and methods." (G) The same as (F) except that the competitive effect of increasing concentration of wild-type AT and AT-N135Q were monitored in presence of fixed concentration of HRPII (40 nM).
AT inhibits HRPII-mediated inflammatory responses in neutrophils
Similar to endothelial cells, HRPII induced activation of NF-κB in dimethyl sulfoxide–differentiated HL60 (dHL60) cells, which are known to exhibit neutrophil-like properties.26,27 Time-course analysis by western blotting of cell lysates indicated that HRPII induces phosphorylation of NF-κB p65 in dHL60 cells after 5 minutes (the first time-point analyzed) (Figure 2A). Simultaneous treatment with both HRPII and AT led to inhibition of NF-κB p65 phosphorylation in dHL60 cells (Figure 2B). Immunofluorescence analysis indicated that HRPII induces extracellular trap formation and histone H3 citrullination (Cit-H3) in dHL60 cells, and AT-WT and AT-N135Q effectively inhibited these proinflammatory responses (Figure 2C). As expected, AT-4Mut exhibited no protective effect in this assay (Figure 2C). Western blot analysis of dHL60 cell lysates confirmed immunofluorescence data, showing the Cit-H3 level is significantly elevated in HRPII-treated cells (Figure 2D). Proinflammatory cytokines PMA and tumor necrosis factor-α (TNF-α), which are known to induce Cit-H3, were used as positive controls in these experiments (Figure 2D). AT effectively inhibited HRPII-mediated elevation of Cit-H3 in dHL60 cells (Figure 2E). AT-N135Q exhibited a higher inhibitory effect, but AT-4Mut failed to block histone Cit-H3 by HRPII in dHL60 cells (Figure 2E).
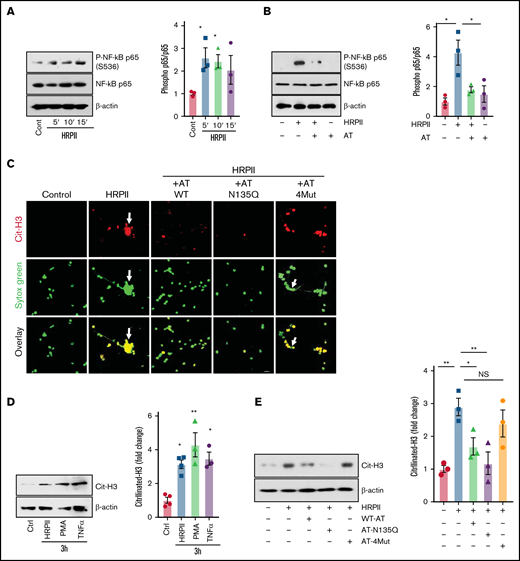
AT inhibits HRPII-mediated inflammatory responses in HL60 cells. (A) dHL60 cells were treated with HRPII (100 nM) for 5 to 15 minutes followed by the analysis of phosphorylation of NF-κB p65 by western blotting. Densitometric analysis is presented. (B) The same as (A) except that cells were treated simultaneously with HRPII and AT (2.5 µM) followed by the analysis of phosphorylation of NF-κB p65 by western blotting. Densitometric analysis is presented. (C) dHL60 cells on coverslips were treated simultaneously with HRPII (100 nM) and different AT derivatives (AT-WT, AT-N135Q and AT-4Mut (2.5 µM) for 4 hours and then fixed in 4% paraformaldehyde, permeabilized with 0.2% TritonX-100/PBS, and stained with anti-citrullinated H3 (rabbit polyclonal) antibody followed by Alexa Fluor 555-conjugated anti-rabbit antibody. DNA was stained with Sytox Green. (D) dHL60 cells were treated with HRPII (100 nM), TNF-α (10 ng/mL), or PMA (100 nM) for 4 hours followed by analysis of histone (H3) citrullination by western blotting. Densitometric analysis is presented. (E) The same as (D) except that cells were treated with HRPII and different AT derivative (AT-WT, AT-N135Q and AT-4Mut (2.5 µM) and histone (H3) citrullination were analyzed by western blotting. Densitometric analysis is presented.
AT inhibits HRPII-mediated inflammatory responses in HL60 cells. (A) dHL60 cells were treated with HRPII (100 nM) for 5 to 15 minutes followed by the analysis of phosphorylation of NF-κB p65 by western blotting. Densitometric analysis is presented. (B) The same as (A) except that cells were treated simultaneously with HRPII and AT (2.5 µM) followed by the analysis of phosphorylation of NF-κB p65 by western blotting. Densitometric analysis is presented. (C) dHL60 cells on coverslips were treated simultaneously with HRPII (100 nM) and different AT derivatives (AT-WT, AT-N135Q and AT-4Mut (2.5 µM) for 4 hours and then fixed in 4% paraformaldehyde, permeabilized with 0.2% TritonX-100/PBS, and stained with anti-citrullinated H3 (rabbit polyclonal) antibody followed by Alexa Fluor 555-conjugated anti-rabbit antibody. DNA was stained with Sytox Green. (D) dHL60 cells were treated with HRPII (100 nM), TNF-α (10 ng/mL), or PMA (100 nM) for 4 hours followed by analysis of histone (H3) citrullination by western blotting. Densitometric analysis is presented. (E) The same as (D) except that cells were treated with HRPII and different AT derivative (AT-WT, AT-N135Q and AT-4Mut (2.5 µM) and histone (H3) citrullination were analyzed by western blotting. Densitometric analysis is presented.
Neutrophil activation and NET formation have been shown to be involved in pathogenesis of inflammation in patients infected with P falciparum.28,29 However, the underlying cause of NET formation in P falciparum infection is not known. We investigated the possible role of HRPII in NET formation using freshly isolated human neutrophils. Results indicated that HRPII induces degranulation of neutrophils, release and colocalization of MPO with extracellular DNA as determined by co-staining for both MPO and Sytox Green (supplemental Figure 2A). Formation of long, extracellular, mesh-like structures of DNA associated with Cit-H3 suggested that HRPII induces NET formation and AT effectively inhibits this response (supplemental Figure 2B). Studies have demonstrated that Cit-H3 plays a key role in mediating NET formation,30 a process in which activated neutrophils release their nuclear contents as decondensed chromatin fibers comprised of DNA, histones, and granular contents to neutralize invading microorganisms.31 Immunofluorescence analysis provided further support for the hypothesis that HRPII induces NET formation by appearance of fiber-like structures co-stained for both neutrophil elastase and Cit-H3 (Figure 3A). Histone citrullination is catalyzed by nuclear peptidyl arginine deiminase 4 (PAD4), which facilitates decondensation of chromatin and formation of NETs by innate immune cells.32,33 Confocal immunofluorescence analysis of neutrophils, co-stained for PAD4/H3/DAPI, indicated that HRPII induces PAD4 nuclear translocation, thereby enhancing citrullination of histones in neutrophils (Figure 3B), providing further support for the hypothesis that HRPII activates neutrophils to induce NETs. HRPII-mediated PAD4 nuclear translocation was further confirmed by western blot analysis of nuclear extract of neutrophils (Figure 3C). In addition to NET release, HRPII also induced secretion of TNF-α, which was also inhibited by AT (Figure 3D). Taken together, these results suggest that AT effectively inhibits HRPII-mediated NET formation and blocks the release of proinflammatory markers in activated neutrophils.
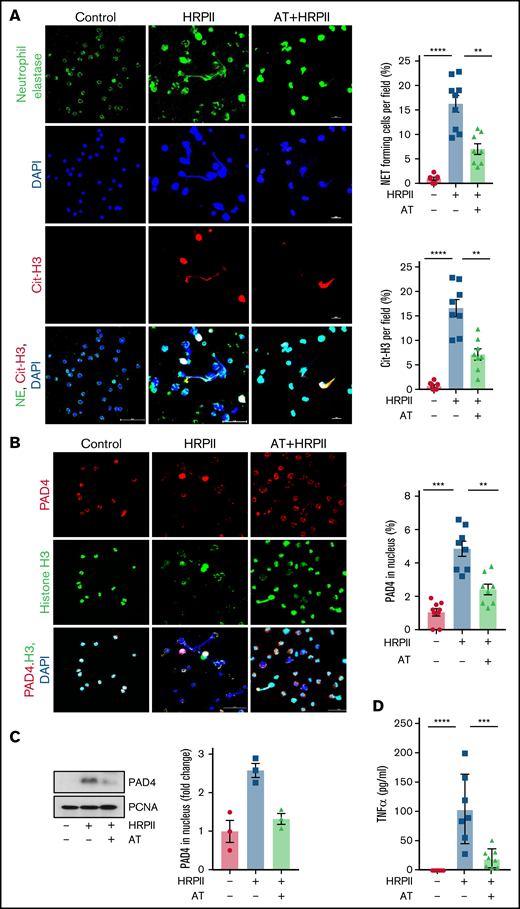
AT inhibits HRPII activation of neutrophils and NET formation. (A) Human blood neutrophils were isolated and treated with HRPII (100 nM) in absence or presence of AT for 4 hours. Cells were then fixed in 4% paraformaldehyde, permeabilized with 0.2% TritonX-100/PBS, and stained with anti-neutrophil elastase (goat polyclonal) antibody, anti-citrullinated H3 (rabbit polyclonal) followed by Alexa Fluor-488 conjugated anti-goat and Alexa Fluor 555-conjugated anti-rabbit antibody. DNA was stained with DAPI. Quantification of NET forming cells and citrullinated H3-positive cells per field represented as the percentage of the total cells. (B) The same as panels above except that human blood neutrophils were fixed and stained with anti-PAD4 (rabbit polyclonal) and antihistone (H3) (mouse monoclonal) antibody followed by Alexa Fluor 555-conjugated anti-rabbit antibody and Alexa Fluor 488-conjugated anti-mouse antibody. DAPI was used to stain the nucleus. Quantification of PAD4 nuclear translocation is represented next to the panel. (C) Human blood neutrophils were treated with HRPII in the absence and presence of AT. PAD4 levels in nuclear extract were analyzed through western blot. PCNA was used as loading control. Densitometric analysis is presented. (D) Human blood neutrophils were treated with HRPII in the absence and presence of AT. TNF-α levels in supernatant of neutrophils were measured using commercial ELISA.
AT inhibits HRPII activation of neutrophils and NET formation. (A) Human blood neutrophils were isolated and treated with HRPII (100 nM) in absence or presence of AT for 4 hours. Cells were then fixed in 4% paraformaldehyde, permeabilized with 0.2% TritonX-100/PBS, and stained with anti-neutrophil elastase (goat polyclonal) antibody, anti-citrullinated H3 (rabbit polyclonal) followed by Alexa Fluor-488 conjugated anti-goat and Alexa Fluor 555-conjugated anti-rabbit antibody. DNA was stained with DAPI. Quantification of NET forming cells and citrullinated H3-positive cells per field represented as the percentage of the total cells. (B) The same as panels above except that human blood neutrophils were fixed and stained with anti-PAD4 (rabbit polyclonal) and antihistone (H3) (mouse monoclonal) antibody followed by Alexa Fluor 555-conjugated anti-rabbit antibody and Alexa Fluor 488-conjugated anti-mouse antibody. DAPI was used to stain the nucleus. Quantification of PAD4 nuclear translocation is represented next to the panel. (C) Human blood neutrophils were treated with HRPII in the absence and presence of AT. PAD4 levels in nuclear extract were analyzed through western blot. PCNA was used as loading control. Densitometric analysis is presented. (D) Human blood neutrophils were treated with HRPII in the absence and presence of AT. TNF-α levels in supernatant of neutrophils were measured using commercial ELISA.
AT attenuates HRPII-mediated inflammation in vivo
The proinflammatory effect of HRPII was evaluated in an in vivo model by IP administration of HRPII in mice. HRPII increased infiltration of MPO-positive neutrophils to lungs, which was significantly attenuated by AT (Figure 4A). Immunofluorescence analysis of lung tissues suggested that number of resident macrophages in lungs (F4/80-positive) does not change (supplemental Figure 3A), suggesting HRPII-mediated infiltrated leukocytes are primarily neutrophils. Ly6G immunostaining provided further support for this hypothesis (supplemental Figure 3B). Analysis of kidney tissues by Ly6G immunostaining yielded similar results, suggesting HRPII also induces neutrophil infiltration to kidneys (supplemental Figure 3C). AT effectively attenuated HRPII-mediated neutrophil infiltration to both lungs and kidneys (supplemental Figure 3B-C). Flow cytometry analysis showed HRPII increases surface expression of Mac1 on blood neutrophils (Figure 4B). HRPII also increased mouse plasma MPO as determined by an antibody-dependent MPO activity assay, and AT significantly suppressed this effect (Figure 4C). Immunofluorescence analysis for Cit-H3 in lung tissues of HRPII-treated mice revealed that infiltrating neutrophils were forming NETs, indicating HRPII is inducing NETs in vivo, and this effect was inhibited by AT (Figure 4D). Western blot analysis of lung tissue lysates showed that AT significantly reduces HRPII-mediated elevation of Cit-H3 and MPO (Figure 4E). These results suggest HRPII activates neutrophils in vivo to promote inflammation, which is attenuated by AT. Western blot analysis further indicated that AT attenuates HRPII-mediated endothelial activation, as measured by expression of CAMs. Expression of ICAM1 and VCAM1 was upregulated in lung tissues of mice treated with HRPII, but it was downregulated in mice treated with both HRPII and AT, as determined by western blot (Figure 5A) and immunofluorescence analyses (supplemental Figure 4A-B), suggesting that HRPII activates lung endothelial cells, thereby promoting infiltration of activated neutrophils in lung tissues, as demonstrated in Figure 4A. In agreement with this observation, HRPII also disrupted barrier-permeability function of lung endothelial cells, and AT inhibited this response (Figure 5B).
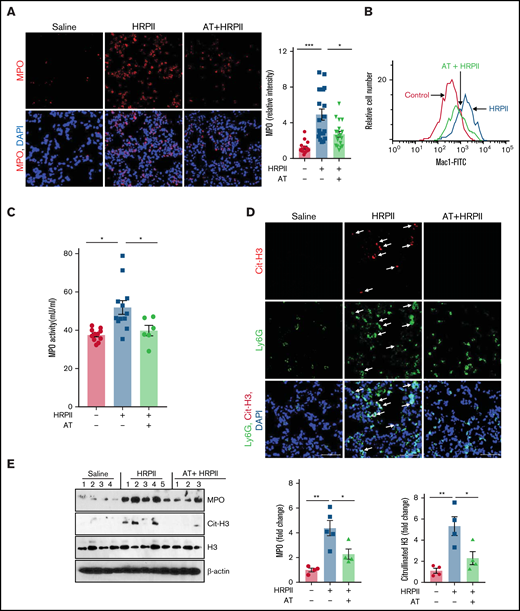
AT inhibits HRPII-mediated activation of neutrophils in vivo. Mice were injected IP with saline or HRPII (10 µg/g) and AT (500 µg/mouse ∼20 µg/g) followed by collection of organs after 3.5 hours for analysis. (A) Cryosections of the lung tissue were fixed and permeabilized followed by analysis of infiltration of neutrophils to the lung with anti-MPO (rabbit) antibody and Alexa Fluor 555-conjugated anti-rabbit antibody. DAPI was used to stain the nucleus. Relative intensity of the MPO stain is presented. (B) Mac1 surface expression in Ly6G-positive mouse neutrophils was analyzed by flow cytometry as described in "Materials and methods." (C) The MPO level in plasma (marker of neutrophil activation) was analyzed by the MPO activity assay as described in "Materials and methods." (D) The same as (A) except that the lung cryosections were fixed, permeabilized, and incubated with anti-citrullinated histone H3 (rabbit) and anti-Ly6G (rat) antibodies followed by Alexa Fluor 555-conjugated anti-rabbit and Alexa Fluor 488-conjugated anti-rat antibodies. DAPI was used to stain the nucleus. The arrows indicate citrullinated histone H3. (E) The PBS-perfused lower right lobe of lung was dissected and dissolved in the tissue lysis buffer followed by western blotting of the lysate for detection of MPO, citrullinated histone H3, histone H3, and β-actin using appropriate antibodies. Densitometric analysis of expression of these proteins is presented.
AT inhibits HRPII-mediated activation of neutrophils in vivo. Mice were injected IP with saline or HRPII (10 µg/g) and AT (500 µg/mouse ∼20 µg/g) followed by collection of organs after 3.5 hours for analysis. (A) Cryosections of the lung tissue were fixed and permeabilized followed by analysis of infiltration of neutrophils to the lung with anti-MPO (rabbit) antibody and Alexa Fluor 555-conjugated anti-rabbit antibody. DAPI was used to stain the nucleus. Relative intensity of the MPO stain is presented. (B) Mac1 surface expression in Ly6G-positive mouse neutrophils was analyzed by flow cytometry as described in "Materials and methods." (C) The MPO level in plasma (marker of neutrophil activation) was analyzed by the MPO activity assay as described in "Materials and methods." (D) The same as (A) except that the lung cryosections were fixed, permeabilized, and incubated with anti-citrullinated histone H3 (rabbit) and anti-Ly6G (rat) antibodies followed by Alexa Fluor 555-conjugated anti-rabbit and Alexa Fluor 488-conjugated anti-rat antibodies. DAPI was used to stain the nucleus. The arrows indicate citrullinated histone H3. (E) The PBS-perfused lower right lobe of lung was dissected and dissolved in the tissue lysis buffer followed by western blotting of the lysate for detection of MPO, citrullinated histone H3, histone H3, and β-actin using appropriate antibodies. Densitometric analysis of expression of these proteins is presented.
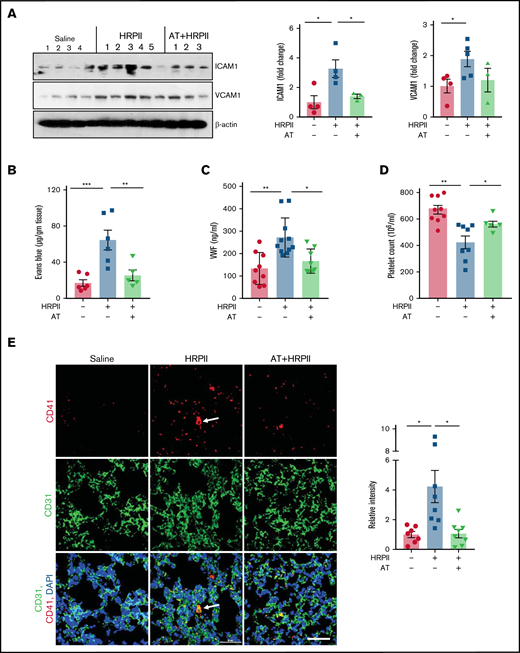
AT inhibits HRPII-mediated inflammation and coagulation. Mice were injected IP with saline or HRPII (10 µg/g) and AT (20 µg/g) followed by euthanizing mice after 3.5 hours, perfusing the lungs with PBS, dissecting the lower right lobe of the lung, and dissolving it in the tissue lysis buffer. (A) Lung tissue lysates were immunoblotted for VCAM1 and ICAM1 and β-actin. Densitometric analysis of expression of these proteins is presented. (B) The barrier-protective effect of AT in mice injected with both AT and HRPII was analyzed after 3 hours by IV injection of 1% Evans blue dye. After 30 minutes, animals were euthanized and perfused with PBS, lung tissue samples were collected, and vascular permeability was measured from the amount of Evans blue dye leaked into the lung as described in "Materials and methods." (C) Plasma level of VWF was measured by ELISA using a commercial kit. (D) Blood platelet counts were determined using a veterinary hematology analyzer. (E) Lung cryosections were fixed, permeabilized, and incubated with anti-CD41 (rat) and anti-CD31 (rabbit) antibodies followed by Alexa Fluor 555-conjugated anti-rat and Alexa Fluor 488-conjugated anti-rabbit antibodies. DAPI was used to stain the nucleus. The arrows indicate platelet-rich thrombus. Relative intensity of CD41 is presented.
AT inhibits HRPII-mediated inflammation and coagulation. Mice were injected IP with saline or HRPII (10 µg/g) and AT (20 µg/g) followed by euthanizing mice after 3.5 hours, perfusing the lungs with PBS, dissecting the lower right lobe of the lung, and dissolving it in the tissue lysis buffer. (A) Lung tissue lysates were immunoblotted for VCAM1 and ICAM1 and β-actin. Densitometric analysis of expression of these proteins is presented. (B) The barrier-protective effect of AT in mice injected with both AT and HRPII was analyzed after 3 hours by IV injection of 1% Evans blue dye. After 30 minutes, animals were euthanized and perfused with PBS, lung tissue samples were collected, and vascular permeability was measured from the amount of Evans blue dye leaked into the lung as described in "Materials and methods." (C) Plasma level of VWF was measured by ELISA using a commercial kit. (D) Blood platelet counts were determined using a veterinary hematology analyzer. (E) Lung cryosections were fixed, permeabilized, and incubated with anti-CD41 (rat) and anti-CD31 (rabbit) antibodies followed by Alexa Fluor 555-conjugated anti-rat and Alexa Fluor 488-conjugated anti-rabbit antibodies. DAPI was used to stain the nucleus. The arrows indicate platelet-rich thrombus. Relative intensity of CD41 is presented.
AT attenuates HRPII-mediated activation of coagulation
Analysis of mice blood samples indicated that HRPII increases circulating plasma levels of VWF and AT effectively inhibits this response (Figure 5C). HRPII also induced thrombocytopenia, as evidenced by lower number of platelets in circulation, which was inhibited by AT (Figure 5D). Immunofluorescence analysis showed markedly high levels of platelets (CD41-positive cells) in both lung (Figure 5E) and kidney (supplemental Figure 5A), indicating increased platelet deposition in both tissues of HRPII-treated mice. These results suggest that HRPII induced VWF release, thrombocytopenia, and platelet deposition, which were all effectively inhibited by AT.
The coagulation activation marker, TAT complex, was also markedly increased in the plasma of HRPII-treated mice (Figure 6A), suggesting that HRPII promotes activation of coagulation and AT inhibits this procoagulant state (Figure 6A). This conclusion was further confirmed by the analysis of fibrin(ogen) deposition in lung tissues. Western blot and immunofluorescence studies showed a markedly elevated level of fibrin(ogen) in lung tissues that was significantly inhibited by AT (Figure 6B-D). Hematoxylin and eosin staining supported this conclusion, as evidenced by appearance of thrombus in lung tissues of HRPII-treated mice, which was again effectively inhibited by AT (Figure 6E). In addition to lungs, HRPII also increased fibrin(ogen) deposition and vascular leakage in the kidney, which were attenuated by AT (supplemental Figure 5B-D). Taken together, increased VWF and TAT levels in blood samples as well as increased platelet and fibrin(ogen) deposition in lung and kidney suggest HRPII can promote activation of coagulation and thrombosis, which are attenuated by AT.
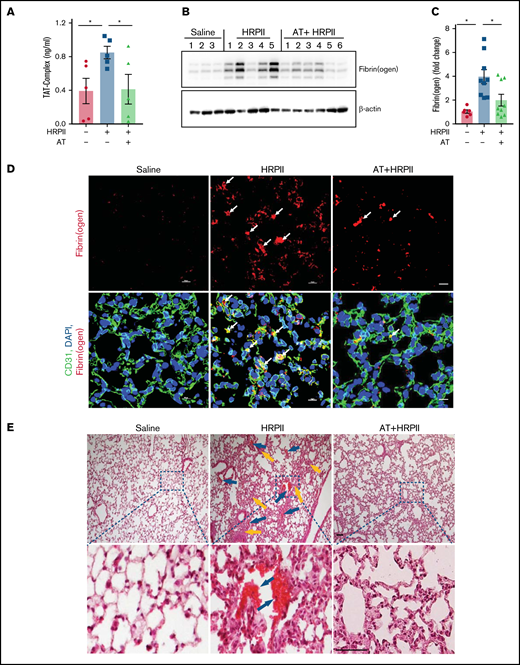
AT inhibits HRPII-mediated procoagulant responses in mice. Mice were injected IP with saline or HRPII (10 µg/g) and AT (20 µg/g) followed by taking blood samples and collecting organs after 3.5 hours for analysis. (A) Plasma level of the TAT complex was measured with established ELISA. (B) Perfused lower right lobe of the lung was harvested in the tissue lysis buffer and immunoblotted for fibrin(ogen). (C) Densitometric analysis of fibrin(ogen) deposition in the lung tissue sample is presented. (D) Lung cryosections were fixed, permeabilized, and incubated with anti-CD31 (rat) and anti-fibrin(ogen) (rabbit) antibodies followed by Alexa Fluor 488-conjugated anti-rat and Alexa Fluor 562-conjugated anti-rabbit antibodies. DAPI was used to stain the nucleus. The arrows indicate intravascular thrombosis. (E) Perfused upper left lobe of the lung was collected and processed for histological analysis. Paraffin-embedded sections of the lung tissue were stained with hematoxylin and eosin. Inset boxes from each group are magnified. Blue arrows indicate thrombosis, and yellow arrows indicate inflammatory foci.
AT inhibits HRPII-mediated procoagulant responses in mice. Mice were injected IP with saline or HRPII (10 µg/g) and AT (20 µg/g) followed by taking blood samples and collecting organs after 3.5 hours for analysis. (A) Plasma level of the TAT complex was measured with established ELISA. (B) Perfused lower right lobe of the lung was harvested in the tissue lysis buffer and immunoblotted for fibrin(ogen). (C) Densitometric analysis of fibrin(ogen) deposition in the lung tissue sample is presented. (D) Lung cryosections were fixed, permeabilized, and incubated with anti-CD31 (rat) and anti-fibrin(ogen) (rabbit) antibodies followed by Alexa Fluor 488-conjugated anti-rat and Alexa Fluor 562-conjugated anti-rabbit antibodies. DAPI was used to stain the nucleus. The arrows indicate intravascular thrombosis. (E) Perfused upper left lobe of the lung was collected and processed for histological analysis. Paraffin-embedded sections of the lung tissue were stained with hematoxylin and eosin. Inset boxes from each group are magnified. Blue arrows indicate thrombosis, and yellow arrows indicate inflammatory foci.
Role of neutrophils on HRPII-mediated activation of inflammation and coagulation
To delineate the role of neutrophils in HRPII-mediated activation of inflammation and coagulation, mice were treated with anti-Ly6G antibody to deplete circulating neutrophils. Proinflammatory effects of HRPII were measured in neutrophil-depleted mice (supplemental Figure 6). Western blot analysis showed that HRPII-mediated upregulation of ICAM1 and VCAM1 was reduced in neutrophil-depleted mice (Figure 7A-C). These results indicate HRPII-mediated neutrophil activation and release of proinflammatory cytokines play an important role in inducing endothelial dysfunction. The MPO level in the lung was also reduced in anti-Ly6G antibody-treated mice (Figure 7A,D); however, fibrin(ogen) deposition was not changed, suggesting that circulating neutrophils may not play a significant role in HRPII-mediated coagulation activation (Figure 7A,E). In support of this hypothesis, plasma levels of TAT complex were also unchanged after neutrophil depletion (Figure 7F). The VWF level in plasma, which was significantly upregulated after HRPII treatment, was reduced after neutrophil depletion (Figure 7G). Circulating platelets are known to bind VWF strings attached to activated endothelial cells. Neutrophil depletion rescued HRPII-mediated thrombocytopenia, supporting a role for neutrophils in activation of endothelial cells and their interaction with platelets (Figure 7H). HRPII-mediated vascular permeability was also significantly decreased in neutrophil-depleted mice (Figure 7I). Taken together, these results suggest neutrophils play an important role in promoting HRPII-mediated proinflammatory signaling but may have a minimal direct role in activation of coagulation.
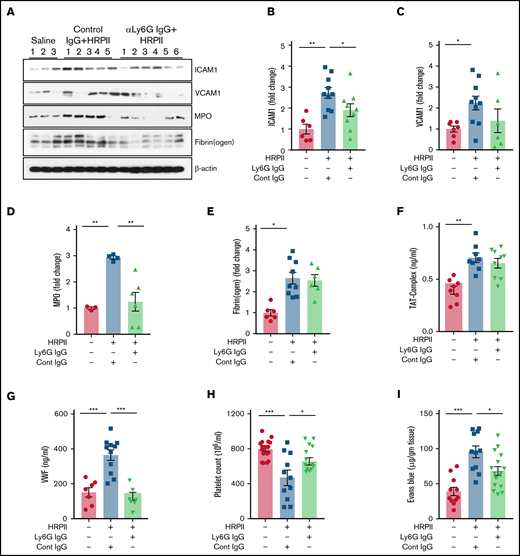
Depletion of neutrophils in mice decreases HRPII-mediated inflammation but not the procoagulant response. Mice were administered either control antibody or anti-Ly6G antibody (20 µg/g) 48 hours before HRPII treatment (10 µg/g). After 3.5 hours of HRPII treatment, mice were euthanized and perfused with PBS, and blood and organs were collected. The lower right lobe of the lung was lysed in tissue lysis buffer. (A) Lung tissue lysates were immunoblotted for VCAM1, ICAM1, MPO, fibrin(ogen), and β-actin. Densitometric analysis of expression of these proteins is presented in (B), (C), (D), and (E). β-actin was used as loading control. (F) Plasma level of the TAT complex was measured with established ELISA. (G) Plasma level of VWF was measured by ELISA using a commercial kit. (H) Blood platelet counts were determined using a veterinary hematology analyzer. (I) To analyze the effect of neutrophil depletion on lung vascular permeability, 1% Evans blue dye was injected 3 hours after HRPII treatment. After 30 minutes, animals were euthanized and perfused with PBS, lung tissue samples were collected, and vascular permeability was measured from the amount of Evans blue dye leaked into the lung as described in “Materials and methods.”
Depletion of neutrophils in mice decreases HRPII-mediated inflammation but not the procoagulant response. Mice were administered either control antibody or anti-Ly6G antibody (20 µg/g) 48 hours before HRPII treatment (10 µg/g). After 3.5 hours of HRPII treatment, mice were euthanized and perfused with PBS, and blood and organs were collected. The lower right lobe of the lung was lysed in tissue lysis buffer. (A) Lung tissue lysates were immunoblotted for VCAM1, ICAM1, MPO, fibrin(ogen), and β-actin. Densitometric analysis of expression of these proteins is presented in (B), (C), (D), and (E). β-actin was used as loading control. (F) Plasma level of the TAT complex was measured with established ELISA. (G) Plasma level of VWF was measured by ELISA using a commercial kit. (H) Blood platelet counts were determined using a veterinary hematology analyzer. (I) To analyze the effect of neutrophil depletion on lung vascular permeability, 1% Evans blue dye was injected 3 hours after HRPII treatment. After 30 minutes, animals were euthanized and perfused with PBS, lung tissue samples were collected, and vascular permeability was measured from the amount of Evans blue dye leaked into the lung as described in “Materials and methods.”
Discussion
We recently demonstrated HRPII binds vascular GAGs to disrupt barrier-permeability function of endothelial cells through Src-dependent phosphorylation and destabilization of VE-cadherin.15 Results here indicate that HRPII, by interacting with GAGs, induces phosphorylation and nuclear translocation of NF-κB p65 and upregulation of ICAM1 in endothelial cells. The essential role of GAGs in proinflammatory function of HRPII can be gleaned from observations that surfen (1) inhibited binding of HRPII to endothelial cells as analyzed by flow cytometry, (2) blocked nuclear translocation of NF-κB p65 as determined by immunofluorescence, and (3) inhibited barrier-disruptive function of HRPII as analyzed by a permeability assay. HRPII and AT bind overlapping sites on endothelial GAGs because simultaneous treatment of cells with both HRPII and AT-WT, but not a signaling-defective d-helix mutant (AT-4Mut), effectively blocked both binding and proinflammatory function of HRPII. The higher affinity GAG variant (AT-N135Q) exhibited significantly higher competitive effect in blocking barrier-disruptive function of HRPII, further supporting the hypothesis that both molecules interact with overlapping binding sites on GAGs to transmit their signaling effects. The molecular mechanism through which HRPII and AT elicit paradoxical signaling effects through interaction with same GAGs is not fully understood. We previously demonstrated that the protective signaling function of AT requires d-helix–dependent interaction of AT with 3-OS containing GAGs,18,34,35 a modification primarily mediated by heparan sulfate 3-O-sulfotransferase-1 (3-OST-1) in endothelial cells.35 The siRNA knockdown of 3-OST-1 abrogated both barrier-protective function of AT and barrier-disruptive function of HRPII,15 further supporting the hypothesis that both molecules competitively bind similar and overlapping GAG sequences to exert their signaling functions. We demonstrated that interaction of AT with 3-OS containing GAGs on syndecan 4 culminates in recruitment of PKC-δ to cytoplasmic membrane of endothelial cells, thereby leading to phosphorylation of syndecan 4 cytoplasmic domain, induction of prostacyclin synthesis, and inhibition of activation of NF-κB.21 However, unlike AT, interaction of HRPII with same type of GAGs resulted in activation of NF-κB by an unknown mechanism. We hypothesize upon interaction with GAGs, as a pathogen-associated molecular pattern, HRPII is likely directed to molecular pattern recognition receptors. A recent study demonstrated that HRPII activates inflammasome to exert proinflammatory effects by a mechanism not dependent on Toll-like receptors (TLR1, TLR2, TLR5, TLR6, and TLR9).13 However, the possible role of receptor for advanced glycation end-products (RAGE) has not been investigated. Previous results have indicated GAGs function as coreceptors for RAGE36 ; thus, the hypothesis that GAG-bound HRPII is presented to RAGE as a possible mechanism to account for proinflammatory function of HRPII warrants further investigation. However, direct proinflammatory signaling by HRPII through interaction with GAGs cannot be excluded at this time.
Similar to activation of endothelial cells, HRPII also activated NF-κB in leukocytes and induced histone citrullination and NET formation in both dHL60 cells and human neutrophils. The nuclear enzyme PAD4 catalyzes histone citrullination, which results in decondensation of chromatin structure and release of nuclear contents as NETs by neutrophils during infection.30-33 Our results suggest that HRPII upregulates expression of PAD4 and its nuclear localization in neutrophils, as demonstrated by confocal imaging. AT inhibited PAD4 expression and NET formation in HRPII-treated neutrophils, possibly by competitively blocking interaction of HRPII with GAGs on neutrophils. In support of this hypothesis, unlike AT-WT, AT-4Mut did not inhibit citrullination of histones and NET formation in dHL60 cells. By contrast, AT-N135Q, which exhibits a higher affinity for GAGs,17,24,25 was more effective in inhibiting these inflammatory processes in HRPII-treated dHL60 cells. Nevertheless, a recent study identified three transmembrane receptors, CD13, CD300f, and LRP-1, on monocytes that bind to AT to initiate protective cell signaling.37 Thus, further studies are required to determine whether these receptors are expressed on neutrophils and whether interaction of AT with any one of these receptors contributes to inhibition of HRPII-mediated citrullination and NET formation by neutrophils. Further studies are also required to understand the mechanism by which HRPII activates neutrophils and, whether similar to interaction with endothelial cells, it interacts with GAGs on neutrophils or its direct interaction with any one of pattern recognition receptors is responsible for transmitting proinflammatory signaling of HRPII. Based on presented results and our experimental approach in this study (simultaneous incubation of cells with HRPII and AT), we hypothesize that similar to endothelial cells, both molecules competitively bind to neutrophil GAGs to exert their specific signaling effects. The observations that AT-4Mut had no effect in protecting neutrophils and AT-N135Q exhibited higher protective activity are consistent with this hypothesis.
The in vivo relevance of HRPII-mediated inflammatory responses was investigated by challenging mice with HRPII in both the presence and absence of AT. Results indicated that HRPII promotes both inflammation and coagulation, and AT exhibits a significant protective effect in downregulating both pathways. This conclusion is derived from observations that HRPII increased cell surface expression of Mac1 on mouse blood neutrophils (Ly6G-positive cells) and promoted their infiltration into lung and kidney tissues. HRPII also upregulated expression of CAMs and vascular permeability in lungs and kidneys. HRPII also induced NET formation in vivo and markedly increased histone Cit-H3 in lung tissues and MPO activity in the plasma. Recent results have indicated a direct association between neutrophil activation and NETosis in malaria severity in patients infected with P falciparum.28,29,38 Thus, it is tempting to speculate that HRPII may contribute to NETosis in patients infected with P falciparum. In support of this possibility, circulating NET counts in patients infected with P falciparum have been found to correlate with the plasma level of HRPII.28 However, the exact mechanism of NET formation in severe malaria needs further investigation because neutrophil activation and NET formation have also been observed in infection with different species of Plasmodium that do not synthesize HRPII.29 In this context, neutrophil activation and NET formation have been shown to be associated with development of acute respiratory distress syndrome in Plasmodium berghei-infected mice, in which neutrophil depletion protects mice from malaria-associated acute respiratory distress syndrome.39 We also found that neutrophil depletion rescues HRPII-mediated endothelial activation, pulmonary vascular leakage, and thrombocytopenia. In addition to promotion of inflammation, HRPII also promoted procoagulant responses, as evidenced by increased platelet deposition in lungs, thrombocytopenia, elevated VWF and TAT complex in the plasma, and fibrin(ogen) in lung tissues. Neutrophil depletion did not have effects on fibrin(ogen) and TAT complex levels, possibly suggesting a minimal direct role for neutrophils in promoting activation of coagulation and thrombin generation. However, neutrophil depletion rescued HRPII-mediated thrombocytopenia and elevated VWF release, both of which are known to play critical roles in pathogenesis of P falciparum malaria.40 The mechanism by which HRPII induces coagulation activation requires further investigation. A prothrombotic role for NETs has been reported by their ability to support platelet adhesion, activation, and aggregation.41 Moreover, NET-associated histones are known to stimulate exocytosis of VWF in endothelial cells and platelets, thereby promoting coagulation and inflammation (immunothrombosis).42 The observation that neutrophil depletion rescued HRPII-mediated thrombocytopenia and elevated VWF release confirms results of these previous studies on the important role of NETs in immunothrombosis; however, the lack of an effect on fibrin(ogen) deposition and TAT complex suggests HRPII also enhances thrombin generation independent of NETs. In this context, in addition to NETs, HRPII also induced TNF-α expression, which may induce tissue factor expression on monocytes and endothelial cells, thereby promoting activation of coagulation. Finally, it is known that activated platelets store significant amounts of polyphosphates, which not only bind HRPII and promote its proinflammatory function15 but also bind histones and promote thrombin generation by a platelet-dependent mechanism.43 Thus, the possibility that activated platelet-derived polyphosphates are involved in augmenting procoagulant function of HRPII also needs further investigation. This is a clinically important question because P falciparum is known to store large amounts of short- and long-chain polyphosphate in different blood stages of infection.44 We have demonstrated that platelet-sized polyphosphates can bind to nuclear proteins and induce VWF release and platelet string formation on endothelial cells.45 Thus, further studies are required to understand the exact mechanism through which HRPII promotes coagulation activation in our model system. Further studies are also required to determine whether HRPII-mediated endothelial cell or platelet activation or both contribute to increased plasma level of VWF in patients with P falciparum malaria.
In summary, our results demonstrate that HRPII promotes inflammation and coagulation in both cellular and animal models; thus, it may play a role in the disease pathogenesis of P falciparum malaria. HRPII exerts its proinflammatory effect through interaction with GAGs. AT exhibits a potent protective effect by eliciting GAG-dependent antiinflammatory responses against HRPII in both systems. Moreover, AT competitively inhibits binding of HRPII to GAGs, thereby downregulating its proinflammatory signaling function. Currently, there is no effective therapy for treating severe malaria. Based on our results, we propose that the therapeutic potential of AT, particularly AT-N135Q, which exhibits significantly higher GAG-binding properties, in protecting against pathogenesis of P falciparum parasite infection may warrant further investigation.
Acknowledgments
The authors thank Cindy Carter for technical assistance and Audrey Rezaie for editorial work on the manuscript.
This work was supported by the National Heart, Lung, and Blood Institute, National Institutes of Health (grants HL101917 and HL062565) (A.R.R.).
Authorship
Contribution: I.B. designed, performed, and analyzed experiments in cellular and animal models; S.R.P. designed, performed, and analyzed all animal and flow cytometry experiments; X.S.C. designed and performed histological analysis and immunofluorescence experiments; H.G. measured the VWF level in the plasma; A.R.R. designed experiments, analyzed data, wrote the manuscript, and supervised the project; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Alireza R. Rezaie, Cardiovascular Biology Research Program, Oklahoma Medical Research Foundation, 825 NE 13th St, Oklahoma City, OK 73104; e-mail: ray-rezaie@omrf.org.

