

Key Points
Safety and efficacy data demonstrated that atezolizumab alone or with azacitidine did not support a favorable risk-benefit profile in MDS.
The differential toxicity profile observed between patients with R/R and those with HMA-naïve MDS requires additional investigation.

Abstract
We present primary results from the phase 1b GO29754 study evaluating the safety and tolerability of atezolizumab, a programmed death-ligand 1 inhibitor, alone and in combination with azacitidine, a hypomethylating agent (HMA), in patients with relapsed/refractory (R/R) or HMA-naïve myelodysplastic syndrome (MDS). Patients with R/R MDS received atezolizumab for 12 months (cohort A) or atezolizumab plus azacitidine for 6 cycles followed by atezolizumab as maintenance for 8 cycles (cohort B). Patients with HMA-naïve MDS received atezolizumab plus azacitidine until loss of clinical benefit (cohort C). Safety, activity, and exploratory end points were investigated. Forty-six patients were enrolled and received treatment (cohort A, n = 11; cohort B, n = 14; cohort C, n = 21). All patients experienced ≥1 adverse event (AE) on study, and all patients discontinued atezolizumab. In cohort A, 7 patients (63.6%) died, and no patients responded. In cohort B, 8 patients (57.1%) discontinued azacitidine, 11 (78.6%) died, and 2 (14.3%) responded. In cohort C, all 21 patients discontinued azacitidine, 13 died (61.9%), and 13 (61.9%) responded. The study was terminated by the sponsor before completion of recruitment because of the unexpected high early death rate in cohort C (6 [46.2%] of 13 deaths were due to AEs and occurred within the first 4 treatment cycles.). The high death rate and poor efficacy observed in this study do not support a favorable risk-benefit profile for atezolizumab as a single agent or in combination with azacitidine in R/R or HMA-naïve MDS. This trial was registered at www.clinicaltrials.gov as #NCT02508870.
Introduction
The myelodysplastic syndromes (MDS) comprise a heterogeneous group of clonal stem cell disorders caused by ineffective hematopoiesis determining a maturation arrest in the bone marrow and pancytopenia in the peripheral blood. MDS predominantly occurs in the elderly, with most patients diagnosed after the age of 60 years.1,2 Low-risk patients (those with a Revised International Prognostic Scoring System [IPSS-R] score of <3.5 points) have a median 4-year survival of 80%, but patients with higher-risk MDS (HR-MDS;IPSS-R ≥ 3.5) have poor prognosis and experience rapid progression, with a median survival of <1 year.3 Standard of care includes supportive care treatments such as blood transfusions or growth factors4 ; patients with HR-MDS also require more intensive treatment, such as hypomethylating agents (HMAs), chemotherapy, and/or allogeneic stem cell transplantation.
Azacitidine, an HMA, improved median overall survival (OS) and delayed progression to acute myeloid leukemia (AML) in elderly patients with HR-MDS in the phase 3 AZA-001 study.5 Despite this clinical benefit, azacitidine therapy is not curative, and patients with HR-MDS who fail to respond to, or relapse/progress after, treatment with an HMA have limited therapeutic options and poor prognosis.6 Therefore, there continues to be a high unmet medical need for new therapies for MDS.
Programmed death-ligand 1 (PD-L1) expression is upregulated in patients with HR-MDS compared with patients with lower-risk MDS7 and those for whom HMA therapy fails,8 and it has been suggested that escape from immune surveillance via overexpression of PD-L1 may play a role in MDS pathogenesis.7 Atezolizumab is a humanized immunoglobulin G1 monoclonal antibody that targets PD-L1 and inhibits the interaction between PD-L1 and its receptors, programmed death-1 (PD-1) and B7-1/CD80.9 Therapeutic blockade of PD-L1 binding by atezolizumab provides antitumor activity in several tumor types.10-13 Combining the inhibition of the PD-L1/PD-1 pathway with azacitidine may offer a potential new therapeutic approach in MDS, with the ability to improve patient outcomes.
We present the primary safety and efficacy results from GO29754, a phase 1b study of atezolizumab as a single agent and in combination with azacitidine in patients with R/R and those with HMA-naïve MDS.
Methods
Patients
Eligible patients had HMA-naïve MDS and were classified by the IPSS-R as intermediate, high, or very high risk or had MDS that had relapsed after, or was refractory to, prior HMA therapy (defined as disease progression [PD] at any time after initiation of azacitidine or decitabine treatment or failure to achieve, or relapse after achieving, complete [CR] or partial response [PR] or hematologic improvement [HI] after at least six 4-week cycles of azacitidine or four 6-week cycles of decitabine administered within the past 2 years). All patients were age ≥ 18 years and had an Eastern Cooperative Oncology Group performance status of 0 to 2, with adequate end-organ function and the ability to comply with the study protocol. Patients were willing and able to undergo pretreatment and subsequent on-treatment bone marrow biopsies.
The study was conducted in accordance with the International Conference on Harmonisation guidelines for Good Clinical Practice, and the protocol was approved by the ethics committees of all participating centers. All patients provided written informed consent.
Study design
GO29754 was a multicenter, open-label, phase 1b study (supplemental Figure 1). Atezolizumab was evaluated as a single agent in the R/R population (cohort A, comprising subgroups A1 and A2) and in combination with azacitidine in the R/R (cohort B, comprising subgroups B1 and B2) and HMA-naïve/frontline (cohort C, comprising subgroups C1 and C2) populations.
In cohort A1, patients received 1200 mg of atezolizumab as an IV infusion once every 3 weeks for 12 months. In cohort B1, patients received 840 mg of atezolizumab IV once every 2 weeks with 75 mg/m2 of azacitidine as a subcutaneous injection on days 1 to 7 every 4 weeks (or on days 1-5 and 8-9 according to institutional preference14 ) for the 6-cycle induction phase, followed by 1200 mg of atezolizumab IV once every 3 weeks for up to 8 cycles (6 months) of maintenance treatment. In cohort C1, patients received 840 mg of atezolizumab IV once every 2 weeks with 75 mg/m2 of azacitidine subcutaneously on days 1 to 7 once every 4 weeks until loss of clinical benefit.
Across all cohorts, if ≤2 patients (of a total of 10 patients in cohort A1, 10 in cohort B1, and 6 in cohort C1) experienced a dose-limiting toxicity within the first cycle of treatment (ie, for cohort A1, within 21 days; for cohorts B1 and C1, within 28 days), then additional patients could be recruited (cohorts A2, B2, and C2). Patients with R/R MDS were randomly assigned to either cohort A2 or B2 at a 1:1 ratio using a stratified permutation block randomization scheme incorporating baseline IPSS-R risk (very low, low, or intermediate vs high or very high) as a stratification factor. Patients with frontline MDS were assigned to cohort C2. Across all cohorts, alternative doses or schedules could be explored if patients experienced unacceptable toxicity.
Study end points
The objectives of the study were to establish the safety and tolerability of atezolizumab monotherapy in patients with HR-MDS who are R/R to HMAs to establish the safety and tolerability of atezolizumab plus azacitidine in patients with HR-MDS who are treatment naïve to HMAs or patients with MDS who are R/R to HMAs and to define the recommended phase 2 dose for atezolizumab plus azacitidine. The primary end point was incidence and nature of dose-limiting toxicities. Other safety end points were incidence, nature, and severity of adverse events (AEs; graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0) and incidence of antidrug antibodies (ADAs) in response to atezolizumab. Secondary end points were pharmacokinetic (PK) and activity outcomes, including overall response rate (ORR; response defined as CR plus marrow CR [mCR] plus PR plus HI according to the 2006 International Working Group Response Criteria for MDS15 ), best objective response (OR; defined as CR, mCR, PR, or HI) achieved during the study, OR at end of induction, duration of response (DOR), time to AML progression (defined as time from first day of study treatment to diagnosis date of AML progression), progression-free survival, OS in patients R/R to HMAs, and changes in red blood cell (RBC) and platelet transfusion rates. Exploratory end points were identification and profiling of biomarkers and patient-reported outcomes.
PK analyses
Atezolizumab serum concentration was measured preinfusion for all cycles and at 30 minutes postinfusion on day 1, cycle 1, for cohort A and on day 8, cycle 1, for cohorts B and C.
Biomarker assessment
Mutation analysis of bone marrow aspirate samples was performed using the FoundationOne Heme platform (Foundation Medicine, Inc., Cambridge, MA) as previously described.12 For RNA sequencing (RNAseq), RNA was isolated and purified from bone marrow mononuclear cells and used to create complementary DNA libraries that were assayed using TruSeq (Illumina, Inc., San Diego, CA) RNA Access (Expression Analysis). RNAseq read counts were converted to log-transformed counts per million using the edgeR package16 for downstream analysis. Phenotypic assessment of leukocyte populations was analyzed centrally by flow cytometry in blood and bone marrow pre- and posttreatment. Lymphocyte subsets (naïve, memory, and regulatory T cells [Tregs]) were determined using CD45, CD3, CD4, CD8, CCR7, and CD45RA (Labcorp, Burlington, NC). T-cell activation and proliferation were measured using major histocompatibility complex 2 cell surface receptor (HLA-DR) and ki67 (Labcorp). PD-L1 cell surface expression was measured on total CD45+ lymphocytes and CD34+ blasts with a proprietary monoclonal antibody (clone 14D3; Genentech, Inc., South San Francisco, CA) directed against a noncompeting PD-L1 epitope (Genentech, Inc.).
Statistical analysis
The intention-to-treat population comprised all enrolled patients. The efficacy- and safety-evaluable populations comprised all patients who received any amount of either study drug. Design considerations covering all cohorts were made not with regard to explicit power or type 1 error considerations but instead to obtain preliminary safety, efficacy, PK, and pharmacodynamic information for atezolizumab as a single agent or in combination with azacitidine. Planned enrollment for this study was ∼100 patients, which depended on the numbers and sizes of the cohorts. Standard descriptive statistics were used to summarize clinical results. Time-to-event data were summarized using the Kaplan-Meier method.
Results
Patient characteristics and treatment
Enrollment began on 30 September 2015. Seventy patients were screened for participation; 46 were enrolled and received treatment (Figure 1). This included 25 patients with R/R MDS (cohort A1, n = 10; cohort A2, n = 1; cohort B1, n = 11; cohort B2, n = 3) and 21 patients receiving frontline treatment (cohort C1, n = 7; cohort C2, n = 14). All 46 patients met the criteria for inclusion in the intention-to-treat, efficacy-evaluable, and safety-evaluable populations.
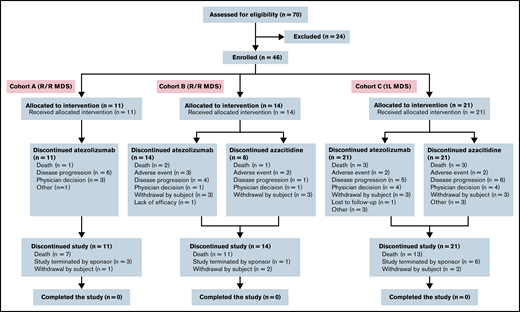
The study was terminated by the sponsor before completion of recruitment because of an unexpected high early death rate observed in cohort C compared with historical controls (last patient last visit and data cutoff date, 11 July 2019).
Baseline demographics
The median age of patients in the R/R cohort was 75 years (range, 63-89), and a majority of patients (17 [68%] of 25) were male (Table 1). Most patients (14 [56%] of 25) had received 1 prior line of therapy. In the frontline cohort, the median age was 72 years (range, 50-81), and a majority of patients (15 [71%] of 21) were male. One patient (from cohort C) had undergone autologous stem cell transplantation.
Patient demographics and baseline disease characteristics (ITT population, R/R cohorts, and 1L cohort)
| Characteristic | R/R MDS | 1L MDS | ||
|---|---|---|---|---|
| Cohort A (n = 11) | Cohort B (n = 14) | All (n = 25) | Cohort C (n = 21) | |
| Age, y | 71 (63-87) | 76 (66-89) | 75 (63-89) | 72 (50-81) |
| Male sex | 8 (72.7) | 9 (64.3) | 17 (68.0) | 15 (71.4) |
| Bone marrow blasts, % | 5 (2-14) | 1 (1-17) | 6 (1-17) | 5 (0-15) |
| Cytogenetics | ||||
| Very good | 0 | 2 (14.3) | 2 (8.0) | 0 |
| Good | 6 (54.5) | 7 (50.0) | 13 (52.0) | 8 (38.1) |
| Intermediate | 1 (9.1) | 4 (28.6) | 5 (20.0) | 8 (38.1) |
| Poor | 1 (9.1) | 0 | 1 (4.0) | 0 |
| Very poor | 3 (27.3) | 1 (7.1) | 4 (16.0) | 5 (23.8) |
| IPSS-R at screening | ||||
| Very low | 0 | 0 | 0 | 0 |
| Low | 0 | 1 (7.1) | 1 (4.0) | 0 |
| Intermediate | 1 (9.1) | 2 (14.3) | 3 (12.0) | 9 (42.9) |
| High | 0 | 0 | 0 | 6 (28.6) |
| Very high | 0 | 0 | 0 | 6 (28.6) |
| Missing/unknown | 10 (90.9) | 11 (78.6) | 21 (84.0) | — |
| ECOG PS | ||||
| 0 | 3 (27.3) | 3 (21.4) | 6 (24.0) | 1 (4.8) |
| 1 | 8 (72.7) | 10 (71.4) | 18 (72.0) | 19 (90.5) |
| 2 | 0 | 1 (7.1) | 1 (4.0) | 1 (4.8) |
| Prior lines of therapy, n | ||||
| 1 | 6 (54.5) | 8 (57.1) | 14 (56.0) | 0 |
| 2 | 3 (27.3) | 1 (7.1) | 4 (16.0) | 0 |
| ≥3 | 2 (18.2) | 5 (35.7) | 7 (28.0) | 1 (4.8) |
| Prior ASCT | 0 | 0 | 0 | 1 (4.8) |
| Characteristic | R/R MDS | 1L MDS | ||
|---|---|---|---|---|
| Cohort A (n = 11) | Cohort B (n = 14) | All (n = 25) | Cohort C (n = 21) | |
| Age, y | 71 (63-87) | 76 (66-89) | 75 (63-89) | 72 (50-81) |
| Male sex | 8 (72.7) | 9 (64.3) | 17 (68.0) | 15 (71.4) |
| Bone marrow blasts, % | 5 (2-14) | 1 (1-17) | 6 (1-17) | 5 (0-15) |
| Cytogenetics | ||||
| Very good | 0 | 2 (14.3) | 2 (8.0) | 0 |
| Good | 6 (54.5) | 7 (50.0) | 13 (52.0) | 8 (38.1) |
| Intermediate | 1 (9.1) | 4 (28.6) | 5 (20.0) | 8 (38.1) |
| Poor | 1 (9.1) | 0 | 1 (4.0) | 0 |
| Very poor | 3 (27.3) | 1 (7.1) | 4 (16.0) | 5 (23.8) |
| IPSS-R at screening | ||||
| Very low | 0 | 0 | 0 | 0 |
| Low | 0 | 1 (7.1) | 1 (4.0) | 0 |
| Intermediate | 1 (9.1) | 2 (14.3) | 3 (12.0) | 9 (42.9) |
| High | 0 | 0 | 0 | 6 (28.6) |
| Very high | 0 | 0 | 0 | 6 (28.6) |
| Missing/unknown | 10 (90.9) | 11 (78.6) | 21 (84.0) | — |
| ECOG PS | ||||
| 0 | 3 (27.3) | 3 (21.4) | 6 (24.0) | 1 (4.8) |
| 1 | 8 (72.7) | 10 (71.4) | 18 (72.0) | 19 (90.5) |
| 2 | 0 | 1 (7.1) | 1 (4.0) | 1 (4.8) |
| Prior lines of therapy, n | ||||
| 1 | 6 (54.5) | 8 (57.1) | 14 (56.0) | 0 |
| 2 | 3 (27.3) | 1 (7.1) | 4 (16.0) | 0 |
| ≥3 | 2 (18.2) | 5 (35.7) | 7 (28.0) | 1 (4.8) |
| Prior ASCT | 0 | 0 | 0 | 1 (4.8) |
Data are presented as n (%) or median (range).
1L, frontline; ASCT, autologous stem cell transplantation; ECOG PS, Eastern Cooperative Oncology Group performance status; ITT, intention to treat.
The median time on study was 6.4 months (range, 2.3-40.5) for patients in cohort A, 10.3 months (range, 0.8-43.9) for those in cohort B, and 18.5 months (range, 0.7-26.5) for those in cohort C.
Safety and tolerability
The median duration of exposure to atezolizumab was 4.2 months (range, 0.8-8.4) in cohort A, 4.6 months (range, 0.0-18.9) in cohort B, and 5.6 months (range, 0.0-25.8) in cohort C. The median duration of exposure to azacitidine was 3.5 months (range, 0.2-5.5) in cohort B and 5.8 months (range, 0.2-25.6) in cohort C.
All patients experienced at least 1 AE on study (Table 2). The most common all-grade AEs (≥40%) were pyrexia (3 [27%] of 11) in cohort A; decreased neutrophil count (7 [50%] of 14), constipation (6 [43%] of 14), and nausea (6 [43%] of 14) in cohort B; and constipation (11 [52%] of 21), diarrhea (10 [48%] of 21), and nausea (9 [43%] of 21) in cohort C.
Summary of AEs (safety-evaluable population, R/R cohorts, and 1L cohort)
| R/R MDS | 1L MDS | ||
|---|---|---|---|
| Cohort A (n = 11) | Cohort B (n = 14) | Cohort C (n = 21) | |
| Patients with at least 1 AE | 11 (100) | 14 (100) | 21 (100) |
| Total events, n | 59 | 186 | 427 |
| Grade 3-4 AE | 5 (45.5) | 13 (92.9) | 18 (85.7) |
| Grade 3-4 AE related to treatment | 1 (9.1) | 10 (71.4) | 13 (61.9) |
| Grade 5 (fatal) AE | 1 (9.1) | 2 (14.3) | 6 (28.6) |
| SAE | 5 (45.5) | 9 (64.3) | 15 (71.4) |
| SAE related to treatment with atezolizumab | 2 (18.2) | 4 (28.6) | 3 (14.3) |
| SAE related to treatment with azacitidine | NA | 5 (35.7) | 5 (23.8) |
| AE leading to treatment modification of atezolizumab | 1 (9.1) | 1 (7.1) | 5 (23.8) |
| AE leading to treatment modification of azacitidine | NA | 7 (50.0) | 3 (14.3) |
| AE leading to discontinuation of atezolizumab | 1 (9.1) | 5 (35.7) | 5 (23.8) |
| AE leading to discontinuation of azacitidine | NA | 3 (21.4) | 5 (23.8) |
| R/R MDS | 1L MDS | ||
|---|---|---|---|
| Cohort A (n = 11) | Cohort B (n = 14) | Cohort C (n = 21) | |
| Patients with at least 1 AE | 11 (100) | 14 (100) | 21 (100) |
| Total events, n | 59 | 186 | 427 |
| Grade 3-4 AE | 5 (45.5) | 13 (92.9) | 18 (85.7) |
| Grade 3-4 AE related to treatment | 1 (9.1) | 10 (71.4) | 13 (61.9) |
| Grade 5 (fatal) AE | 1 (9.1) | 2 (14.3) | 6 (28.6) |
| SAE | 5 (45.5) | 9 (64.3) | 15 (71.4) |
| SAE related to treatment with atezolizumab | 2 (18.2) | 4 (28.6) | 3 (14.3) |
| SAE related to treatment with azacitidine | NA | 5 (35.7) | 5 (23.8) |
| AE leading to treatment modification of atezolizumab | 1 (9.1) | 1 (7.1) | 5 (23.8) |
| AE leading to treatment modification of azacitidine | NA | 7 (50.0) | 3 (14.3) |
| AE leading to discontinuation of atezolizumab | 1 (9.1) | 5 (35.7) | 5 (23.8) |
| AE leading to discontinuation of azacitidine | NA | 3 (21.4) | 5 (23.8) |
Data presented as n (%).
1L, frontline; NA, not applicable; SAE, serious AE.
Grade ≥3 AEs were reported in 46% (5 of 11) of patients in cohort A, 93% (13 of 14) of patients in cohort B, and 91% (19 of 21) of patients in cohort C. No grade ≥3 AE was reported in >1 patient in cohort A. The most commonly reported grade ≥3 AEs (≥20%) in cohort B were neutrophil count decreased (6 [43%] of 14), febrile neutropenia (5 [36%] of 14), and neutropenia (3 [21%] of 14). The most commonly reported grade ≥3 AEs (≥20%) in cohort C were neutropenia (8 [38%] of 21) and febrile neutropenia (7 [33%] of 21). Serious AEs were reported in 46% (5 of 11) of patients in cohort A, 64% of patients in cohort B, and 71% of patients in cohort C.
AEs of special interest to atezolizumab were reported in 2 patients (18%) in cohort A (immune-mediated rash and infusion-related reaction [IRR]) and 9 patients (64%) in cohort B (comprising most commonly immune-mediated rash, immune-mediated hepatitis, and IRR; supplemental Table 1). A majority of events were grade 1 or 2 in severity (grade 3: cohort A, 9.1%; cohort B, 21%); no event grade >3 in severity was reported. AEs of special interest to atezolizumab were reported in 9 patients (43%) in cohort C (comprising most commonly immune-mediated rash and immune-mediated hepatitis; supplemental Table 2). A majority were grade 1 or 2 in severity (grade 3, 10%).
Four patients received systemic corticosteroid treatment for immune-mediated AEs. Three patients (cohort A, n = 2; cohort C, n = 1) experienced an IRR that manifested as a rash, 1 of which (cohort A) was deemed related to atezolizumab; all resolved with treatment, and no dose interruption/modification was necessary. The fourth patient (cohort B) experienced grade 3 pneumonitis deemed related to atezolizumab; atezolizumab was discontinued on study day 463. The patient died as a result of hypokalemia on study day 764.
Most patients discontinued treatment; all 25 patients in the R/R cohorts (cohorts A and B) and all 21 patients in the frontline cohort (cohort C) discontinued atezolizumab, and 8 of 14 patients in cohort B and all 21 patients in cohort C discontinued azacitidine. The most common reasons for discontinuation included PD, death, AEs, and physician decision (Table 3).
Reasons for withdrawal/discontinuation of treatment
| Atezolizumab | Azacitidine | |||||
|---|---|---|---|---|---|---|
| R/R MDS | 1L MDS | R/R MDS | 1L MDS | |||
| Cohort A (n = 11) | Cohort B (n = 14) | Cohort C (n = 21) | Cohort A (n = 11) | Cohort B (n = 14) | Cohort C (n = 21) | |
| Received at least 1 dose of study treatment | 11 (100) | 14 (100) | 21 (100) | NA | 14 (100) | 21 (100) |
| Withdrawn from treatment | 11 (100) | 14 (100) | 21 (100) | NA | 8 (57.1) | 21 (100) |
| Reason for discontinuation | ||||||
| Death | 1 (9.1) | 2 (14.3) | 3 (14.3) | NA | 1 (7.1) | 3 (14.3) |
| AE | 0 | 3 (21.4) | 2 (9.5) | NA | 2 (14.3) | 2 (9.5) |
| PD | 6 (54.5) | 4 (28.6) | 5 (23.8) | NA | 1 (7.1) | 6 (28.6) |
| Physician decision | 3 (27.3) | 1 (7.1) | 4 (19.0) | NA | 1 (7.1) | 4 (19.0) |
| Withdrawal by patient | 0 | 3 (21.4) | 3 (14.3) | NA | 3 (21.4) | 3 (14.3) |
| Lack of efficacy | 0 | 1 (7.1) | 0 | NA | 0 | 0 |
| Lost to follow-up | 0 | 0 | 1 (4.8) | NA | 0 | 0 |
| Other | 1 (9.1) | 0 | 3 (14.3) | NA | 0 | 3 (14.3) |
| Atezolizumab | Azacitidine | |||||
|---|---|---|---|---|---|---|
| R/R MDS | 1L MDS | R/R MDS | 1L MDS | |||
| Cohort A (n = 11) | Cohort B (n = 14) | Cohort C (n = 21) | Cohort A (n = 11) | Cohort B (n = 14) | Cohort C (n = 21) | |
| Received at least 1 dose of study treatment | 11 (100) | 14 (100) | 21 (100) | NA | 14 (100) | 21 (100) |
| Withdrawn from treatment | 11 (100) | 14 (100) | 21 (100) | NA | 8 (57.1) | 21 (100) |
| Reason for discontinuation | ||||||
| Death | 1 (9.1) | 2 (14.3) | 3 (14.3) | NA | 1 (7.1) | 3 (14.3) |
| AE | 0 | 3 (21.4) | 2 (9.5) | NA | 2 (14.3) | 2 (9.5) |
| PD | 6 (54.5) | 4 (28.6) | 5 (23.8) | NA | 1 (7.1) | 6 (28.6) |
| Physician decision | 3 (27.3) | 1 (7.1) | 4 (19.0) | NA | 1 (7.1) | 4 (19.0) |
| Withdrawal by patient | 0 | 3 (21.4) | 3 (14.3) | NA | 3 (21.4) | 3 (14.3) |
| Lack of efficacy | 0 | 1 (7.1) | 0 | NA | 0 | 0 |
| Lost to follow-up | 0 | 0 | 1 (4.8) | NA | 0 | 0 |
| Other | 1 (9.1) | 0 | 3 (14.3) | NA | 0 | 3 (14.3) |
1L, frontline; NA, not applicable.
A total of 18 patients (72%) with R/R MDS died (cohort A, 7 [64%] of 11; cohort B, 11 [79%] of 14): 3 (17%) as a result of an AE deemed by the investigator to be unrelated to atezolizumab or azacitidine (AE of unexplained cause of death, n = 2; sepsis, n = 1), 5 (28%) as a result of PD, and 6 as a result of unknown causes; the remaining 4 did not report a clear cause of death but had ongoing AEs of hypokalemia, respiratory distress, pneumonia, and septic arthritic knee at the time of death. Thirteen patients in the frontline cohort (61.9%) died. Of these, 6 patients (46.2%) died within the first 4 treatment cycles, with 2 within the first treatment cycle (AE of unexplained cause of death, sepsis, small intestinal obstruction, and device-related infection, n = 1 each; multiorgan dysfunction syndrome, n = 2). Five patients (39%) in this cohort died as a result of PD, and the remaining 2 had an unclear cause of death reported, with ongoing cardiorespiratory issues and hematoma.
Efficacy
There were no responses seen in cohort A; responses were reported in 2 patients (14.3%) in cohort B (CR, 14%) and 13 patients (62%) in cohort C (CR, 19%; mCR, 19%; mCR + HI, 10%; HI, 14%; Table 4). The median DOR was 7.4 months (95% confidence interval [CI], 2.8-12.0) in cohort B and 19.0 months (95% CI, 3.8-21.9) in cohort C (Figure 2). The median time to AML progression was 4.9 months (95% CI, 2.7-8.9) in cohort A, not estimable in cohort B, and 21.7 months (95% CI, 11.6-21.7) in cohort C.
Summary of efficacy (R/R cohorts and 1L cohort)
| R/R MDS | 1L MDS | ||
|---|---|---|---|
| Cohort A (n = 11) | Cohort B (n = 14) | Cohort C (n = 21) | |
| ORR (CR, PR, mCR, HI, mCR + HI), n (%) | 0 | 2 (14.3) | 13 (61.9) |
| HRR (CR, PR, HI, mCR + HI), n (%) | 0 | 2 (14.3) | 9 (42.9) |
| DOR, d (95% CI) | NE | 224.5 (84.0-365.0) | 577.0 (116.0-668.0) |
| Median time to AML progression, d (range) | 148 (39-271) | NE | 662.0 (1.0-662.0) |
| Median PFS, d | 138 | 240 | 309 |
| Median OS, d | 265 | 361 | 659 |
| R/R MDS | 1L MDS | ||
|---|---|---|---|
| Cohort A (n = 11) | Cohort B (n = 14) | Cohort C (n = 21) | |
| ORR (CR, PR, mCR, HI, mCR + HI), n (%) | 0 | 2 (14.3) | 13 (61.9) |
| HRR (CR, PR, HI, mCR + HI), n (%) | 0 | 2 (14.3) | 9 (42.9) |
| DOR, d (95% CI) | NE | 224.5 (84.0-365.0) | 577.0 (116.0-668.0) |
| Median time to AML progression, d (range) | 148 (39-271) | NE | 662.0 (1.0-662.0) |
| Median PFS, d | 138 | 240 | 309 |
| Median OS, d | 265 | 361 | 659 |
1L, frontline; HRR, hematologic response rate; NE, not estimable; PFS, progression-free survival.
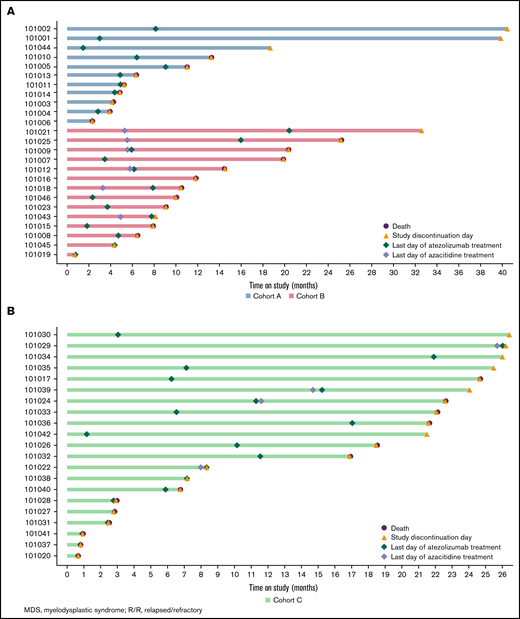
Swimmer plots of time on study. Patients with R/R MDS in cohorts A and B (A) and patients with frontline MDS in cohort C (B).
Swimmer plots of time on study. Patients with R/R MDS in cohorts A and B (A) and patients with frontline MDS in cohort C (B).
At the time of the analysis, the proportion of patients who had progressed or died was 82% in cohort A, 79% in cohort B, and 67% in cohort C. The median progression-free survival was 4.5 months (95% CI, 2.7-8.9) in cohort A, 7.9 months (95% CI, 2.9-11.9) in cohort B, and 10.2 months (95% CI, 3.0-21.7) in cohort C. The median OS was 8.7 months (95% CI, 4.8 to not estimable) in cohort A, 11.9 months (95% CI, 9.1-20.3) in cohort B, and 21.7 months (95% CI, 6.8-22.6) in cohort C.
Of the 7 patients who were transfusion dependent at baseline in cohort A, 5 experienced a decrease (range, −81% to −7%) and 2 an increase (17% and 18%) from baseline in RBC transfusion frequency. Of the 7 transfusion-dependent patients in cohort B, 3 experienced a decrease (range, −69% to −17%) and 4 an increase (range, 5% to 791%) from baseline in RBC transfusion frequency. Of the 12 transfusion-dependent patients in cohort C, 7 experienced a decrease (range, −97% to 16%) and 5 an increase (range, 5% to 536%) from baseline in RBC transfusion frequency. All remaining patients were RBC transfusion independent before the study and transfusion dependent at the end of the study. No patient in cohorts A or B and 2 patients in cohort C experienced a decrease from baseline in platelet transfusion frequency (−63% and −7%). Four patients in cohort A (range, 100% to 173%), 2 in cohort B (49% and 86%), and 4 in cohort C (range, 123% to 479%) experienced an increase from baseline in platelet transfusion frequency. Four patients in cohort A, 4 in cohort B, and 3 in cohort C were platelet transfusion independent before the study and remained so throughout. Twelve patients (cohort A, n = 1; cohort B, n = 5; cohort C, n = 6) became transfusion dependent during the study. Only 1 patient (cohort C) was transfusion dependent before the study but was transfusion independent at the end.
PKs, pharmacodynamics, and immunogenicity
PK analyses were performed pre- and postatezolizumab infusion to determine maximum and minimum serum concentrations (data not shown).
Atezolizumab treatment was previously reported to induce a transient increase in CD8+ki67+HLA-DR+ T cells in peripheral blood,17 and this was therefore assessed as a pharmacodynamic marker. A threefold or greater transient increase in CD8+ki67+HLA-DR+ cells was observed after cycle 1 in cohort B (3 [60%] of 5) and cohort C (7 [64%] of 11). No data were available for cohort A. Notably, this pharmacodynamic effect was not associated with clinical response (supplemental Figure 2).
No atezolizumab-treated patients had a positive result for ADAs at baseline. The postbaseline treatment-emergent ADA incidence was 36% for all dose groups (supplemental Table 3). Because the study was terminated early, analyses of the impact of ADAs on safety, efficacy, or PKs were not performed.
Biomarker assessment
Mutation analysis demonstrated that efficacy was not associated with any specific mutation profile. Notably, several patients with TP53 mutation achieved a response (supplemental Figure 3).
To explore immune correlates within this study, we evaluated PD-L1 expression, CD4 and CD8 effector memory cells, and Tregs by both flow cytometry and RNAseq in bone marrow aspirates. Expression of PD-L1 at baseline was not associated with clinical response by either RNAseq in bulk samples or flow cytometry in gated CD34+ blasts and lymphocyte populations; however, some unfavorable trends were observed in patients with frontline MDS (Figure 3; supplemental Figure 4). Expression of PD-L1 by RNAseq in bulk samples and by flow cytometry in gated CD34+ blasts was significantly associated with inferior OS assessed by univariate Cox regression (supplemental Table 4; supplemental Figure 4).
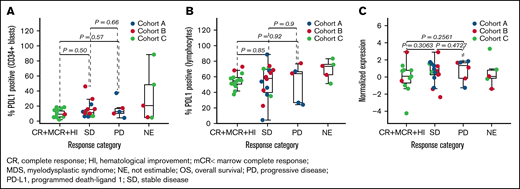
Association of response with PD-L1. PD-L1 protein assayed by flow cytometry in CD34+ blasts (A), PD-L1 protein assayed by flow cytometry in lymphocytes (B), and PD-L1 gene (CD274) expression assayed by RNAseq (C).
Association of response with PD-L1. PD-L1 protein assayed by flow cytometry in CD34+ blasts (A), PD-L1 protein assayed by flow cytometry in lymphocytes (B), and PD-L1 gene (CD274) expression assayed by RNAseq (C).
Increased Tregs either showed a trend toward association or were significantly associated with poor response and inferior OS, especially in frontline patients (supplemental Figure 5). Greater numbers of CD4+ and CD8+ T effector memory cells also showed a trend toward association with inferior outcomes in frontline patients (supplemental Figure 6).
Discussion
In the current study, the safety of atezolizumab monotherapy in patients with HMA-naïve HR-MDS was shown to be largely consistent with its expected profile. However, the combination of atezolizumab plus azacitidine in HMA-naïve patients was associated with high mortality rates, which led to early study termination. Furthermore, atezolizumab alone or in combination with azacitidine had limited clinical activity in patients with MDS previously exposed to HMAs, although this was without excessive or unexpected toxicity.
Previous studies have shown that single-agent immune checkpoint inhibitors demonstrate some limited efficacy in patients with MDS. For example, a phase 1b trial of pembrolizumab (PD-1 inhibitor) monotherapy in 27 patients with HR-MDS who had previously been exposed to HMAs demonstrated an ORR of 3.7% (1 patient with PR).18 A similar phase 1 study of ipilimumab (anti-CTLA4 antibody) monotherapy in 29 patients with HR-MDS demonstrated a safe toxicity profile but limited efficacy, with an ORR of just 3.4% (1 patient with mCR).19 However, it has been shown that the combination of an immune checkpoint inhibitor with an HMA can be effective and tolerable. For example, in an early report of a phase 2 study of nivolumab (PD-1 inhibitor) plus azacitidine in 20 patients with HMA-naïve MDS, overall response was observed in 75% of patients, with CR/CR with incomplete platelet recovery observed in 50%20 (comparable to the overall response observed in HMA-naive patients from cohort C in the current study [13 (62%) of 21]). The 1-year survival rate in these patients was double that in patients for whom HMAs failed (n = 15) who received nivolumab as monotherapy (50% vs 25%, respectively); the authors also concluded that the combination of nivolumab plus azacitidine demonstrated an acceptable toxicity profile.
Patients with adverse cytogenetic risk have previously been shown to have short-lived responses to HMAs compared with patients with normal karyotype.21 For example, poor-risk cytogenetics have been shown to independently predict poor OS in patients with HR-MDS receiving azacitidine (P = .03).22 In addition, poor performance status, high transfusion burden, and presence of peripheral blasts have also been identified as prognostic clinical markers of poorer outcomes in patients receiving HMAs.21,22 Baseline characteristics of patients in the R/R and frontline cohorts in the current study were broadly comparable, although some features correlating with poor outcome were heterogeneously distributed; for example, fewer patients in the frontline cohort had very good or good cytogenetic risk compared with patients in the R/R cohort (38% vs 66%, respectively). These disparities may in part explain the differing safety profile observed for these 2 patient groups, with patients receiving frontline treatment in cohort C experiencing much higher rates of both AEs and serious AEs than R/R patients in cohorts A and B.
More deaths in the frontline cohort (cohort C) were attributed to AEs (46%) compared with deaths in the R/R cohorts (cohort A, 14%; cohort B, 18%). Although there seems to be no clear biologic mechanism that fully explains the additional toxicity of the atezolizumab plus azacitidine combination in the frontline cohort, the fact that antibiotic prophylaxis was left to investigator discretion may have led to an increased infection risk across all cohorts. In addition, the median duration of exposure to azacitidine was longer, and the median number of azacitidine doses was higher, for patients in cohort C (frontline) compared with patients in cohort B (R/R; 5.8 months and 40 doses vs 3.5 months and 27 doses, respectively). However, it should be noted that the patient number in each cohort was small, making it difficult to draw firm conclusions from these data.
High numbers of patients in both the frontline and R/R cohorts experienced AEs that led to azacitidine dose modification or interruption (52% and 71%, respectively). The most commonly reported AE was neutropenia. This is broadly in line with previous studies, such as the phase 3 AZA-001 trial of azacitidine in patients with HR-MDS, in which 66% of patients experienced neutropenia.5 An analysis of AZA-001 and CALGB 9221, a similar phase 3 study of azacitidine in patients with MDS, observed that both hematologic and nonhematologic AEs decreased in frequency as treatment continued and that they could be mostly managed by supportive care measures and dose delays/reductions, thus allowing patients to continue therapy.23 In the current study, the incidence of febrile neutropenia was higher in patients receiving atezolizumab plus azacitidine (cohort B, 36%; cohort C, 33%) vs those receiving atezolizumab alone (cohort A, 9%). Growth factors (eg, granulocyte colony-stimulating factor) have previously been used to mitigate neutropenia related to treatment with HMAs in patients with MDS24 ; however, evidence supporting their use is limited, and in the current study, use of these factors was left to investigator discretion, with no mandatory reporting.
In our study, patients in the frontline cohort were much more likely to withdraw from azacitidine treatment because of PD than patients in the R/R cohorts (28.6% vs 7.1% of patients, respectively). However, patients in the frontline cohort demonstrated more favorable efficacy outcomes compared with patients in the R/R cohorts, with 13 patients (61.9%) in cohort C achieving an OR compared with no patient in cohort A and 2 patients (14.3%) in cohort B. The median DOR and time to AML progression were also prolonged in patients in cohort C compared with those in cohorts A and B. These data suggest that the atezolizumab plus azacitidine combination potentially demonstrates higher efficacy in the frontline population than in the R/R population, although the poor safety outcomes in this cohort confound interpretation of these results, and further study is needed to understand this paradox.
Biomarker analysis demonstrated that PD-L1 expression at baseline and transient increases in T-cell activation and proliferation were not associated with clinical response. However, expression of PD-L1 at baseline (by RNAseq in bulk samples and by flow cytometry in gated CD34+ blasts but not lymphocytes) was significantly associated with inferior OS. The lack of PD-L1 association with response but significant association with OS suggests PD-L1 expression may be prognostic in MDS. The associations found between increased Tregs and greater numbers of CD4+ and CD8+ T effector memory cells and inferior outcomes suggest that immunosuppression may be a significant obstacle in treating MDS with checkpoint inhibitors, particularly in frontline patients, and warrant further evaluation.
In conclusion, the safety findings and the limited efficacy data generated by this study did not support a favorable risk-benefit profile for atezolizumab alone or in combination with azacitidine in patients with R/R and HMA-naïve MDS and supported the decision not to further develop this treatment in this indication. Better understanding of the reasons associated with the differential toxicity profile observed between HMA-naïve vs HMA-failure patients with HR-MDS would be crucial for any potential future developments of similar combinations.
Acknowledgments
This trial was sponsored by F. Hoffmann-La Roche, Ltd. Third-party medical writing assistance, under the direction of the authors, was provided by Helen Cathro, Ashfield MedComms, an Ashfield Health company, and was funded by F. Hoffmann-La Roche, Ltd.
Authorship
Contribution: All authors were involved in the writing, review, and/or revision of the manuscript; W.D., A.T.G., T.L.L., B.L.S., M.D., B.C.M., M.Y., K.Y., C.G., and C.M. were involved with the analysis and interpretation of the data; A.T.G., K.Y., M.Y., Y.F., C.G., and M.D. were involved in the development of the methodology; A.T.G. and B.C.M. were involved in administrative, material, or technical support; W.D., A.T.G., P.G., D.A.P., B.L.S., A.V., M.D., B.C.M., and C.M. were involved in the acquisition of the data; and A.T.G., B.L.S., M.D., B.C.M., and T.L.L. were involved in study supervision.
Conflict-of-interest disclosure: A.T.G. reports consultancy (PharmaEssentia, Constellation, Bristol-Myers Squibb, Sierra Oncology, and AbbVie). T.L.L. reports research funding (Tolero Pharmaceuticals, Gilead Sciences, Prescient Therapeutics, Ono Pharmaceutical, Bio-Path Holdings, Roche/Genentech, Celator, Trovagene, AbbVie, Pfizer, Celgene, Novartis, Astellas Pharma, Seattle Genetics, Incyte, Celyad, and Aptevo Therapeutics [all payments made to the institution]). W.D. reports consultancy (AbbVie, Amgen, PTC Therapeutics, and Seattle Genetics [all payments made to the institution]) and research funding (AbbVie, Aileron Therapeutics, AstexPharmaceuticals, AstraZeneca, BellicumPharmaceuticals, Bristol-Myers Squibb, Celgene, CTI Biopharma, Forma Therapeutics, Forty Seven, Genentech, Inc., H3 Biomedicine, Incyte, Janssen, KaryopharmTherapeutics, Kite Pharma, MedImmune, Pfizer, PTC Therapeutics, Daiichi Sankyo, Seattle Genetics, Takeda, TCR2 Therapeutics, Celularity, and Ryvu Therapeutics [all payments made to the institution]). D.A.P. reports consultancy (Janssen, Forty Seven, Amgen, Genentech, Novartis, Karyopharm, Syndax, Syros, AbbVie, Daiichi Sankyo, Takeda, Pfizer, Celgene/Bristol-Myers Squibb, and Agios), research funding (AbbVie), and other relationships (Glycomimetics). A.V. reports research funding (Bristol-Myers Squibb, GlaxoSmithKline, Incyte, Medpacto, Curis, and Eli Lilly), scientific advising (Stelexis, Novartis, Acceleron, and Celgene), and equity holding (Stelexis and Clinstreet). M.D. is an employee of Genentech, Inc., and owns stock in Roche. C.M., C.G., B.C.M., and Y.F. are employees of Genentech, Inc. M.Y. and K.Y. are employees of F. Hoffman-La Roche, Ltd. P.G. and B.L.S. declare no competing financial interests.
Correspondence: Aaron T. Gerds, Taussig Cancer Institute, Cleveland Clinic, Cleveland, OH 44106; e-mail: gerdsa@ccf.org.

