

Key Points
Immunochemotherapy does not preclude adequate antibody responses in hematology patients, except when B cells are absent or dysfunctional.
During the pandemic, COVID-19 vaccination should not be delayed in hematology patients.

Abstract
Vaccination guidelines for patients treated for hematological diseases are typically conservative. Given their high risk for severe COVID-19, it is important to identify those patients that benefit from vaccination. We prospectively quantified serum immunoglobulin G (IgG) antibodies to spike subunit 1 (S1) antigens during and after 2-dose mRNA-1273 (Spikevax/Moderna) vaccination in hematology patients. Obtaining S1 IgG ≥ 300 binding antibody units (BAUs)/mL was considered adequate as it represents the lower level of S1 IgG concentration obtained in healthy individuals, and it correlates with potent virus neutralization. Selected patients (n = 723) were severely immunocompromised owing to their disease or treatment thereof. Nevertheless, >50% of patients obtained S1 IgG ≥ 300 BAUs/mL after 2-dose mRNA-1273. All patients with sickle cell disease or chronic myeloid leukemia obtained adequate antibody concentrations. Around 70% of patients with chronic graft-versus-host disease (cGVHD), multiple myeloma, or untreated chronic lymphocytic leukemia (CLL) obtained S1 IgG ≥ 300 BAUs/mL. Ruxolitinib or hypomethylating therapy but not high-dose chemotherapy blunted responses in myeloid malignancies. Responses in patients with lymphoma, patients with CLL on ibrutinib, and chimeric antigen receptor T-cell recipients were low. The minimal time interval after autologous hematopoietic cell transplantation (HCT) to reach adequate concentrations was <2 months for multiple myeloma, 8 months for lymphoma, and 4 to 6 months after allogeneic HCT. Serum IgG4, absolute B- and natural killer–cell number, and number of immunosuppressants predicted S1 IgG ≥ 300 BAUs/mL. Hematology patients on chemotherapy, shortly after HCT, or with cGVHD should not be precluded from vaccination. This trial was registered at Netherlands Trial Register as #NL9553.
Introduction
Since the outbreak of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, it has become apparent that patients with hematologic diseases are at the highest risk for severe COVID-19, COVID-19–related death, and persistent viral shedding.1-4 In a large population-based study including >17 million residents of England, it was demonstrated that the mortality risk of SARS-CoV-2 infection in patients diagnosed with a hematologic malignancy in the past 5 years was >2.5-fold higher compared with individuals who had never been diagnosed with a malignant hematologic condition.1 Mortality in patients with sickle cell disease was 5% compared with 0% in a matched cohort of health care professionals.2 In early 2021, the Dutch government decided to prioritize COVID-19 vaccination in immunocompromised individuals, including patients with hematologic conditions. Given the pressure on the health care system at that time in the pandemic and the urgent need to protect as many individuals as possible, it was decided to vaccinate all patients, irrespective of current disease or treatment status.
This allowed us to prospectively measure the immunogenicity of the mRNA-1273 (Moderna/Spikevax) vaccination in those patients that are generally considered too immunocompromised to benefit from vaccination and in whom vaccinations are often postponed until (later) after treatment, such as patients currently receiving, or just after (immuno-)chemotherapy, early after transplantation, or after CD19 chimeric antigen receptor (CAR) T-cell therapy, and in patients receiving novel, targeted therapies.5,6 By quantifying antibody concentrations against the World Health Organization (WHO) standard,7 we were able to identify those patients that despite their underlying condition mounted an adequate antibody response after mRNA-1273 vaccination. Our results demonstrate that in the midst of the pandemic, COVID-19 vaccination should not be postponed in hematology patients, even when under active or shortly after (immuno-)chemotherapy or cell therapy.
Methods
Study participants
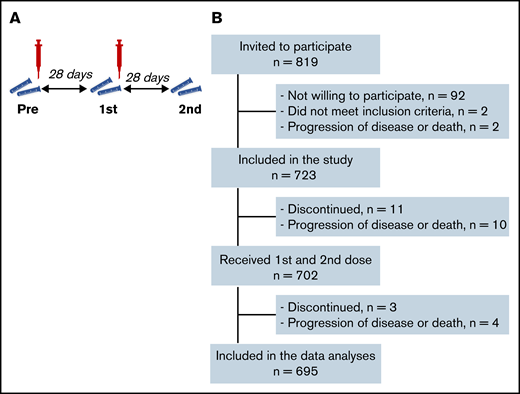
In this prospective, observational multicenter cohort study, we included 723 adult hematology patients, classified into 17 different predefined cohorts based on their diagnosis and treatment status (Table 1; supplemental Table 1). Patient cohorts were defined based on relevance for the hematology practice (eg, disease incidence [chronic lymphocytic leukemia (CLL), multiple myeloma] and knowledge gap [less often studied agents in the context of vaccination or often excluded treatment groups]), with the aim to cover benign, lymphoid, and myeloid diseases. Inclusion criteria were age ≥18 years, a diagnosis of lymphoma, multiple myeloma, CLL, acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), myeloproliferative disease, sickle cell disease, and receiving immunochemotherapy or having received such therapy preferably 6 months but maximally 12 months prior to vaccination, or received targeted agents, or have received autologous or allogeneic hematopoietic cell transplantation (HCT) preferably 6 months but maximally 12 months prior to vaccination, or having received CD19-directed CAR T-cell therapy. All these patients are considered immunocompromised, which is also reflected by their increased risk of COVID-19–related death.1 Exclusion criteria included unwilling or unable to give informed consent, known allergy to one of the components of the vaccine, and a life expectancy of <12 months. Prior infection with SARS-CoV-2 was not an exclusion criterion because the expected seroprevalence was <5% (because of the stringent isolation measures in this patient population), and a test-first strategy for seroprevalence would seriously hamper the speed of vaccination rollout, whereas vaccination of seropositive patients was indicated nonetheless, according to the national vaccination guidelines. All participants received 2 doses of mRNA-1273 28 days apart (Figure 1A). Study protocols were approved by the Institutional Review Board of the Amsterdam UMC and participating centers. All patients provided written informed consent prior to study onset.
Baseline characteristics stratified by cohorts
| n | Age | Sex | WHO PS | Previous SARS-CoV-2* | ||
|---|---|---|---|---|---|---|
| Mean (SD) | Women (%) | 0 to 1, n (%) | 2 to 3, n (%) | n (%) | ||
| All patients | 723 | 59 (12) | 286 (39.6) | 686 (95.4) | 33 (4.6) | 34 (4.9) |
| Sickle cell disease | ||||||
| Hydrea | 31 | 38 (12) | 14 (5.2) | 30 (96.7) | 1 (3.2) | 3 (10.3) |
| Lymphoma | ||||||
| During rituximab±chemotherapy | 46 | 59 (13) | 20 (43.5) | 44 (97.7) | 1 (2.2) | 1 (2.3) |
| <12 mo after rituximab±chemotherapy | 40 | 62 (11) | 17 (42.5) | 38 (95.0) | 2 (5.0) | 0 (0.0) |
| <12 mo after autologous HCT (BEAM)†,‡ | 31 | 58 (12) | 11 (35.5) | 28 (90.3) | 3 (9.7) | 0 (0.0) |
| Multiple myeloma | ||||||
| First-line therapy | 28 | 62 (7) | 12 (42.9) | 26 (92.9) | 2 (7.1) | 1 (3.7) |
| Daratumumab | 52 | 63 (8) | 19 (36.5) | 49 (94.3) | 3 (5.8) | 1 (2.0) |
| IMiDs | 55 | 60 (9) | 21 (38.2) | 54 (98.2) | 1 (1.8) | 6 (10.9) |
| <9 mo after autologous HCT (high-dose melphalan)† | 51 | 61 (7) | 17 (33.3) | 48 (94.1) | 3 (5.9) | 5 (10.0) |
| CLL | ||||||
| Watch and wait | 56 | 65 (8) | 27 (48.2) | 56 (100) | 0 (0) | 3 (5.8) |
| Ibrutinib§ | 38 | 63 (8) | 13 (34.2) | 38 (100 | 0 (0) | 2 (5.3) |
| CML | ||||||
| TKI | 52 | 54 (13) | 26 (50.0) | 51 (98.1) | 1 (1.9) | 2 (3.8) |
| AML and high-risk MDS | ||||||
| Hypomethylating therapy‖ | 19 | 66 (13) | 4 (21.1) | 16 (84.2) | 3 (15.8) | 0 (0.0) |
| High-dose chemotherapy† | 22 | 50 (16) | 11 (50.0) | 20 (90.9) | 2 (9.1) | 1 (5.3) |
| Myeloproliferative disease | ||||||
| Ruxolitinib | 38 | 57 (10) | 16 (42.1) | 38 (100) | 0 (0) | 3 (8.1) |
| Allogeneic HCT | ||||||
| <6 mo after HCT† | 54 | 55 (13) | 21 (38.9) | 48 (88.9) | 6 (11.1) | 1 (2.0) |
| cGVHD | 57 | 57 (10) | 20 (35.1) | 51 (91.0) | 5 (8.9) | 3 (5.7) |
| CAR T-cell therapy | ||||||
| CD19-directed† | 53 | 60 (11) | 17 (32.1) | 53 (100) | 0 (0) | 2 (3.9) |
| n | Age | Sex | WHO PS | Previous SARS-CoV-2* | ||
|---|---|---|---|---|---|---|
| Mean (SD) | Women (%) | 0 to 1, n (%) | 2 to 3, n (%) | n (%) | ||
| All patients | 723 | 59 (12) | 286 (39.6) | 686 (95.4) | 33 (4.6) | 34 (4.9) |
| Sickle cell disease | ||||||
| Hydrea | 31 | 38 (12) | 14 (5.2) | 30 (96.7) | 1 (3.2) | 3 (10.3) |
| Lymphoma | ||||||
| During rituximab±chemotherapy | 46 | 59 (13) | 20 (43.5) | 44 (97.7) | 1 (2.2) | 1 (2.3) |
| <12 mo after rituximab±chemotherapy | 40 | 62 (11) | 17 (42.5) | 38 (95.0) | 2 (5.0) | 0 (0.0) |
| <12 mo after autologous HCT (BEAM)†,‡ | 31 | 58 (12) | 11 (35.5) | 28 (90.3) | 3 (9.7) | 0 (0.0) |
| Multiple myeloma | ||||||
| First-line therapy | 28 | 62 (7) | 12 (42.9) | 26 (92.9) | 2 (7.1) | 1 (3.7) |
| Daratumumab | 52 | 63 (8) | 19 (36.5) | 49 (94.3) | 3 (5.8) | 1 (2.0) |
| IMiDs | 55 | 60 (9) | 21 (38.2) | 54 (98.2) | 1 (1.8) | 6 (10.9) |
| <9 mo after autologous HCT (high-dose melphalan)† | 51 | 61 (7) | 17 (33.3) | 48 (94.1) | 3 (5.9) | 5 (10.0) |
| CLL | ||||||
| Watch and wait | 56 | 65 (8) | 27 (48.2) | 56 (100) | 0 (0) | 3 (5.8) |
| Ibrutinib§ | 38 | 63 (8) | 13 (34.2) | 38 (100 | 0 (0) | 2 (5.3) |
| CML | ||||||
| TKI | 52 | 54 (13) | 26 (50.0) | 51 (98.1) | 1 (1.9) | 2 (3.8) |
| AML and high-risk MDS | ||||||
| Hypomethylating therapy‖ | 19 | 66 (13) | 4 (21.1) | 16 (84.2) | 3 (15.8) | 0 (0.0) |
| High-dose chemotherapy† | 22 | 50 (16) | 11 (50.0) | 20 (90.9) | 2 (9.1) | 1 (5.3) |
| Myeloproliferative disease | ||||||
| Ruxolitinib | 38 | 57 (10) | 16 (42.1) | 38 (100) | 0 (0) | 3 (8.1) |
| Allogeneic HCT | ||||||
| <6 mo after HCT† | 54 | 55 (13) | 21 (38.9) | 48 (88.9) | 6 (11.1) | 1 (2.0) |
| cGVHD | 57 | 57 (10) | 20 (35.1) | 51 (91.0) | 5 (8.9) | 3 (5.7) |
| CAR T-cell therapy | ||||||
| CD19-directed† | 53 | 60 (11) | 17 (32.1) | 53 (100) | 0 (0) | 2 (3.9) |
WHO PS, World Health Organization Performance Status.
N IgG ≥ 14.3 BAUs/mL at baseline.
See supplemental Table 1 for clinical details.
B-NHL: n = 21; Hodgkin lymphoma: n = 2; T-NHL: n = 8.
Plus venetoclax: n = 8; plus obinutuzumab: n = 2.
Hypomethylating therapy: azacitidine (n = 10), azacitidine plus venetoclax (n = 4), decitabine (n = 4), and decitabine plus midostaurine (n = 1).
Clinical parameters
Prevaccination baseline leukocyte count, humoral and cellular immune parameters, including quantitative assessment of lymphocytes, B-, T-, and natural killer (NK)-cell numbers, and serum immunoglobulin M (IgM), IgG, and IgG subtypes 1 to 4, demographic parameters, and medical history, including comorbidities and concomitant medication, were collected prospectively prior to each vaccination and 28 days after the second vaccination (Figure 1A). Clinical data were collected via standardized case report forms.
Antibody concentrations, definitions, and neutralization
Humoral responses (IgG) against subunit 1 (S1), receptor binding domain (RBD), and nucleocapsid (N) antigen domains of SARS-CoV-2 were determined quantitatively and centrally, using a bead-based multiplex immune assay.8 SARS-CoV-2 binding antibody concentration was calibrated against the first serum standard for COVID-19 (20/136), as provided by the National Institute for Biological Standards Control, also recommended by the WHO to define specific serum antibody concentrations in an international perspective.7-9 Seroconversion was defined as obtaining an S1 IgG concentration >10 binding antibody units (BAUs)/mL, and an adequate vaccine response as S1 IgG ≥ 300 BAUs/mL, the IgG concentration that met a plaque reduction neutralization titer 50 of 40 or higher in 2 independent prospective Dutch mRNA-1273 vaccination cohorts.10-13 Reference antibody levels were extracted from the PIENTER cohort,14 a nationwide surveillance cohort with >3000 randomly selected Dutch citizens between 2 and 90 years of age that periodically provide a self-collected fingerstick blood sample and complete an online questionnaire. From this cohort, we selected all age-matched participants who had received a second dose of mRNA-1273 14 to 61 days prior to blood sampling (supplemental Table 2). To test antibody neutralization activity, we generated lentiviral-based pseudoviruses expressing the SARS-CoV-2 wild-type variant (D614G) and determined the 50% inhibitory dose as the dilution of plasma with a 50% reduction of infection using HEK293T-ACE2 cells in all patients with 50 to 300 S1 IgG BAUs/mL and in a random selection of patients with S1 IgG ≥ 300 BAUs/mL (supplemental Table 3).15 SARS-CoV-2 pseudovirus neutralization correlated significantly with authentic SARS-CoV-2 neutralization as measured by plaque reduction neutralization titer or virus neutralization test.15
Statistical analysis
We aimed to include n = 50 patients per subgroup to estimate S1 IgG ≥ 300 BAUs/mL titers with a precision between 7% (at 0 or 100%) and 29% (at 50%) for Clopper-Pearson (exact) 95% confidence intervals (CI). Differences between groups and timepoints were analyzed with Mann-Whitney U and Wilcoxon signed-rank tests, respectively. Univariable and multivariable logistic regression models were used to identify factors associated with S1 IgG ≥ 300 BAUs/mL 4 weeks after the second vaccination. Factors tested include prevaccination baseline leukocyte count, humoral and cellular immune parameters including lymphocyte, B-, T-, and NK-cell numbers, serum IgM, IgG, and IgG subtypes 1 to 4, demographic parameters, and immunosuppressive therapies. Multivariable models were built including all patients irrespective of cohort using a stepwise forward selection procedure (P value for entry .05) with all univariable significant demographic, therapy-related, and immunologic parameters, first as part of their subcategory of parameter, followed by a multivariable analysis of all significant parameters together. A sensitivity analysis was performed for the last model by excluding patients with untreated CLL. Pearson correlation was calculated between serum S1 IgG and in vitro pseudovirus neutralization after 10log-transformation of both. Analyses with 2-sided P values of <.05 were considered statistically significant. Statistical analyses were performed using the IBM SPSS Statistics for Windows, Version 26.0 (IBM Corp, Armonk, NY) and R for Windows, Version 4.0.3 (The R Foundation for Statistical Computing, Vienna, Austria). Normality of continuous parameters was assessed by visual inspection of QQ plots. The linearity assumption for continuous parameters in the logistic regression models was investigated by dividing the parameter in quartiles, and parameters violating the linearity assumption were categorized in clinically relevant groups. The aim of this observational study is to identify those patients that respond sufficiently to COVID-19 vaccination. Because of the observational and descriptive nature of this study, there was no formal hypothesis testing framework.
Results
SARS-CoV-2 antibodies and neutralization
A total of 723 patients were included, 695 of whom were available for analysis after completion of the vaccination schedule (Figure 1B; Table 1). Thirty-one study participants had sickle cell disease and were using hydroxyurea. All other participants had been diagnosed with a hematologic malignancy and were receiving or had recently received targeted therapy, tyrosine kinase inhibitor (TKI) treatment, hypomethylating therapy, (immune)chemotherapy with or without HCT, or CD19-directed CAR T-cell therapy. Median S1 and RBD binding antibody concentrations increased significantly after 2 mRNA-1273 vaccinations, but overall remained significantly lower than the reference population (Figure 2A; supplemental Figure 1). A minority of patients (4.9%) had N-specific IgG > 14.3 BAUs/mL at baseline, indicative of previous SARS-CoV-2 infection (Table 1; supplemental Table 4). As they were primed for SARS-CoV-2 before vaccination, they had better S1 IgG responses than previously uninfected patients and obtained median S1 IgG concentrations comparable to those obtained in healthy individuals (Figure 2A). They were excluded from further analyses. The majority (70%) of previously uninfected participants reached seroconversion (S1 IgG > 10 BAUs/mL), and 55% obtained S1 IgG ≥ 300 BAUs/mL (Figure 2A; supplemental Table 5). Thirty percent of previously uninfected patients did not seroconvert at all. The correlation between serum S1 IgG and in vitro pseudovirus neutralization was strong (r = 0.85; P < .001; Figure 2B). In the 5 months after completion of the vaccination schedule (from 28 days after the second vaccination onwards), 17 (out of 723) patients tested SARS-CoV-2 positive. Seven of these patients received antibody therapy, either convalescent plasma (n = 2) or casirivimab/imdevimab (n = 5). All 17 patients experienced symptomatic infection, and 3 patients required hospital care, 2 of which at the ICU. All 3 hospitalized patients were nonresponders (S1 IgG < 10 BAUs/mL). None of the study participants died because of COVID-19.
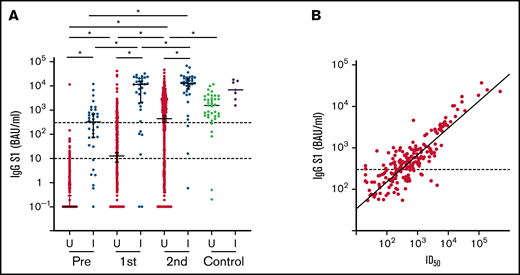
S1 binding antibody concentration and neutralization. (A) IgG S1 concentration for each timepoint. Red and green: previously uninfected patients (U); blue and purple: previously infected patients (I). Dotted lines indicate seroconversion (S1 IgG > 10 BAUs/mL) and sufficient S1 IgG concentration (≥300 BAUs/mL). *P < .05. (B) Correlation of IgG S1 concentration and pseudovirus neutralization (r = 0.85; P < .001). ID50, 50% inhibitory dose.
S1 binding antibody concentration and neutralization. (A) IgG S1 concentration for each timepoint. Red and green: previously uninfected patients (U); blue and purple: previously infected patients (I). Dotted lines indicate seroconversion (S1 IgG > 10 BAUs/mL) and sufficient S1 IgG concentration (≥300 BAUs/mL). *P < .05. (B) Correlation of IgG S1 concentration and pseudovirus neutralization (r = 0.85; P < .001). ID50, 50% inhibitory dose.
Differences between patient cohorts
Median S1 IgG increased significantly in all cohorts after 2 doses of mRNA-1273 (Figure 3). An antibody concentration ≥300 BAUs/mL was obtained in 96% of patients with sickle cell disease on hydroxyurea therapy, in 100% of patients with CML on TKI therapy, and in 94% of patients with AML or high-risk MDS on or <12 months after high-dose chemotherapy (Figure 3; supplemental Table 5). S1 IgG ≥ 300 BAUs/mL was also observed in more than half of patients with multiple myeloma on first-line remission-induction therapy (52%), on daratumumab-containing therapy (69%), on immunomodulatory imide drugs (IMiD; 77%), or within 9 months after high-dose melphalan and autologous HCT (89%), in patients with untreated CLL (70%), and in allogeneic HCT recipients with chronic graft-versus-host disease (cGVHD; 69%). Less than half of patients with myeloproliferative disease on ruxolitinib (49%), with AML or high-risk MDS on hypomethylating therapy (41%), with lymphoma early after BEAM (carmustine, etoposide, cytarabine, melphalan) and autologous HCT (33%), patients shortly after allogeneic HCT (33%), and patients with CLL on ibrutinib (27%) obtained S1 IgG ≥ 300 BAUs/mL. Only few patients with B-non-Hodgkin lymphoma (NHL) on (0%) or within 12 months after (26%) rituximab monotherapy or rituximab-containing immunochemotherapy, and of CD19-directed CAR T-cell therapy recipients (11%) obtained S1 IgG ≥ 300 BAUs/mL (Figure 3; supplemental Table 5). The few responding patients in the CAR T-cell cohort all had normal B-cell numbers. Individual RBD binding antibody concentrations showed similar dynamics over time (not shown).
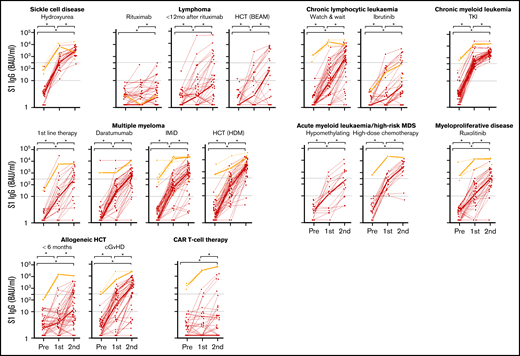
IgG S1 concentration over time for each patient. Thin lines depict previously uninfected patients (red) and previously infected patients (orange); thick lines indicate the median IgG S1 concentrations. Dotted lines specify seroconversion (S1 IgG > 10 BAUs/mL) and sufficient S1 IgG concentration (≥300 BAUs/mL). *P < .05. BEAM, carmustine, etoposide, cytarabine, melphalan.
IgG S1 concentration over time for each patient. Thin lines depict previously uninfected patients (red) and previously infected patients (orange); thick lines indicate the median IgG S1 concentrations. Dotted lines specify seroconversion (S1 IgG > 10 BAUs/mL) and sufficient S1 IgG concentration (≥300 BAUs/mL). *P < .05. BEAM, carmustine, etoposide, cytarabine, melphalan.
Factors associated with antibody responses
Of the immunologic factors, demographic parameters and immunosuppressive medications identified to be associated with obtaining S1 IgG ≥ 300 BAUs/mL in the univariable models, several remained significant in a multivariable model per subcategory of variables (Figure 4A) and in an overall model (Figure 4B; supplemental Table 6). In the overall model, and across cohorts, age correlated significantly with vaccination response (odds ratio [OR], 0.87 per 5-year increase; 95% CI, 0.79-0.96; P = .005). IgG4 concentration (OR, 1.8 per g/L; CI, 1.1-3.1; P = .019) and absolute number of NK cells higher than the upper limit of normal (OR, 8.4; CI, 2.9-24.4; P < .001) were associated with obtaining S1 IgG ≥ 300 BAUs/mL. Lower than normal numbers of B cells (<0.1 × 109/L) decreased odds to obtain S1 IgG ≥ 300 BAUs/mL (OR, 0.25; CI, 0.15-0.42; P < .001). Higher than normal number of B cells (>0.5 × 109/L) was also associated with decreased odds to obtain S1 IgG ≥ 300 BAUs/mL (OR, 0.35; CI, 0.18-0.67; P < .001), but this association was lost when patients with CLL were excluded (OR, 1.15; CI, 0.40-3.28; supplemental Table 6). Absolute CD3 and CD8 T-cell numbers were significantly associated with obtaining S1 IgG ≥ 300 BAUs/mL when tested per subcategory of parameters but not in the overall model. The use of rituximab (OR, 0.24; CI, 0.10-0.57; P = .001), the Bcl2 inhibitor venetoclax (OR, 0.11; CI, 0.019-0.63; P = .013), and CD19-directed CAR T-cell therapy (OR, 0.16; CI, 0.057-0.48; P = .001) was negatively associated with obtaining S1 IgG ≥ 300 BAUs/mL. This also applied to the number of immunosuppressants used (1-2 immunosuppressants: OR, 0.45; CI, 0.28-0.73; >2: OR, 0.25; CI, 0.10-0.64; P = .001)). The use of specific immunosuppressants, such as mycophenolic acid, calcineurin inhibitors, corticosteroids, or the jak2 inhibitor ruxolitinib, was not significantly associated with the odds to obtain S1 IgG ≥ 300 BAUs/mL whether analyzed in the overall model or per patient cohort (allogeneic HCT, cGVHD, and CLL on ibrutinib; supplemental Table 7). Lenalidomide or pomalidomide treatment and the use of TKI on the other hand were associated with higher odds to obtain S1 IgG ≥ 300 BAUs/mL (OR, 3.5; CI, 1.8-6.7; P < .001 and OR, 2.7; CI, 1.1-7.1; P = .038, respectively; Figure 4B).
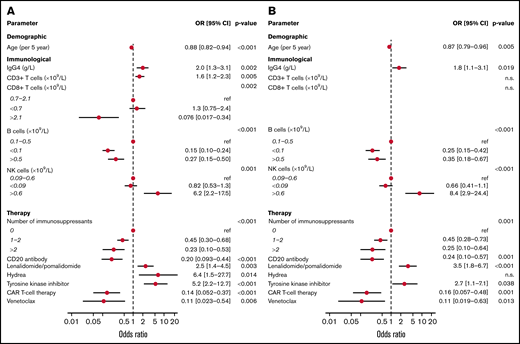
Multivariable analyses of factors associated with S1 IgG responses. (A) Variables significantly associated with S1 IgG ≥ 300 BAUs/mL as a dichotomous outcome tested in a multivariate model per subcategory of variables. (B) Significant variables of model 1 tested in an overall model.
Multivariable analyses of factors associated with S1 IgG responses. (A) Variables significantly associated with S1 IgG ≥ 300 BAUs/mL as a dichotomous outcome tested in a multivariate model per subcategory of variables. (B) Significant variables of model 1 tested in an overall model.
Within cohorts, time after cessation of rituximab, after autologous HCT, or after allogeneic HCT was not significantly associated with the odds to obtain sufficient S1 IgG concentrations (lymphoma: P = .069; multiple myeloma: P = .98; allogeneic HCT: P = .272; supplemental Table 7). Nevertheless, a time interval after which sufficient S1 IgG concentrations were reached could be identified for each cohort. In patients with lymphoma, median S1 IgG ≥ 300 BAUs/mL were not reached before 8 months after cessation of rituximab therapy and after autologous HCT (Figure 5A), whereas in patients with multiple myeloma sufficient S1 IgG concentrations were obtained immediately after autologous HCT (Figure 5B). In allogeneic HCT recipients, a median S1 IgG ≥ 300 BAUs/mL was not reached before 4 to 6 months after transplantation (Figure 5C). In all groups, numbers of circulating B cells did not need to be normalized to obtain S1 IgG ≥ 300 BAUs/mL (Figure 5).
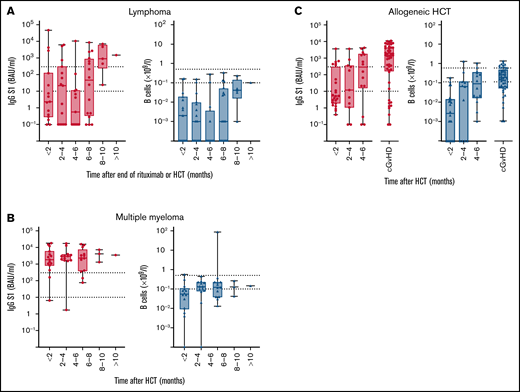
IgG S1 concentration and B cell numbers related to time after end of rituximab or HCT. (A) Patients with lymphoma after autologous HCT or after rituximab. (B) Multiple myeloma after autologous HCT. (C) Allogeneic HCT. Left panels: S1 IgG concentration with dotted lines specifying seroconversion (S1 IgG > 10 BAUs/mL) and sufficient S1 IgG concentration (≥300 BAUs/mL). Right panels: B-cell number with dotted lines at upper and lower limit of normal.
IgG S1 concentration and B cell numbers related to time after end of rituximab or HCT. (A) Patients with lymphoma after autologous HCT or after rituximab. (B) Multiple myeloma after autologous HCT. (C) Allogeneic HCT. Left panels: S1 IgG concentration with dotted lines specifying seroconversion (S1 IgG > 10 BAUs/mL) and sufficient S1 IgG concentration (≥300 BAUs/mL). Right panels: B-cell number with dotted lines at upper and lower limit of normal.
Discussion
At the time COVID-19 vaccines became available, vaccination guidelines in hematology patients under active treatment were conservative, owing to the paucity of vaccine response data and the many uncertainties regarding the immunocompetence of these patients.5,6 Our study is unique in that it was designed to specifically include severely immunocompromised hematology patients during or shortly after treatment or HCT, for whom vaccinations are often deemed futile. Moreover, we performed a quantitative analysis of SARS-CoV-2 binding antibody concentrations, calibrated against the WHO standard,7,10-13,15 to identify those patients who despite their immunodeficiency did obtain adequate antibody concentrations. In collaboration with a number of other prospective cohort studies on the immunogenicity of COVID-19 vaccination among immunocompromised and healthy individuals conducted in The Netherlands, we set an S1 IgG concentration of 300 BAUs/mL as the lower threshold of an adequate COVID-19 vaccine response.9,11,13-16 It is interesting to note that a lower threshold of 264 BAUs/mL was very recently, in an independent British cohort, estimated to correspond to a vaccine efficacy against symptomatic COVID-19 of 80%.7 Even though we included only patients considered to be severely immunocompromised, 70% demonstrated significant increases in antibody concentrations, and >50% obtained adequate antibody concentrations. In particular, patients with sickle cell disease, CML on TKI, AML, and high-risk MDS on or <12 months after high-dose chemotherapy, multiple myeloma on IMiD or daratumumab therapy, patients with untreated CLL and patients with cGVHD had higher odds to obtain adequate S1 IgG concentrations than anticipated. Serum S1 IgG correlated strongly with in vitro antibody-mediated virus neutralization, confirming data obtained from studies in healthy individuals, patients with kidney disease, and oncology patients.11,13,17,18
In general, S1 IgG responses were higher in our study than reported previously. For example, seroconversion was reached in 83% of untreated patients with CLL, compared with 55% in a cohort of therapy-naive patients with CLL vaccinated with BNT162b2.19 Apart from the differences in vaccination schedule and age, which was lower in our cohort (median age 64 vs 71 years), type of vaccine is a possible determining factor. All participants in our study received mRNA-1273, which induces higher antibody concentrations than other vaccines.20-23 Whether this corresponds to better protection against severe COVID-19 remains to be determined.
Vaccination guidelines for autologous HCT recipients do not typically differentiate between patient groups.5 Our data demonstrate, however, that vaccine immunogenicity after autologous HCT depends on the underlying hematologic condition and/or treatment. In patients with multiple myeloma, adequate antibody responses were reached immediately after HCT, whereas patients with lymphoma did not obtain adequate S1 IgG titers before at least 8 months after HCT, similar as observed in patients with B-NHL after cessation of rituximab-containing therapy. These differences are probably related to differences in the degree of B-cell depletion between cohorts, that was much less stringent in patients with multiple myeloma. Of note, our results demonstrate that also low numbers of B cells can produce adequate levels of antibodies. In multivariable analyses, IgG4 concentration and absolute B- and NK-cell numbers predicted S1 IgG responses. Not only lower-than-normal B-cell numbers were associated with blunted antibody responses, but also higher-than-normal B-cell numbers reduced odds to obtain sufficient S1 IgG concentrations. As this correlation was no longer significant when patients with CLL were excluded from the analysis, this is most likely related to CLL-associated reduced numbers of normal B cells and/or immune paralysis.24 SARS-CoV-2 vaccination responses were better in patients using the immunomodulator lenalidomide, as demonstrated previously for pneumococcal vaccination,25 and in patients with CML using TKI. The latter is in contrast to previous reports demonstrating reduced immunogenicity of pneumococcal vaccination during TKI therapy,26 which may be related to the distinctive pathways of immunity induced by messenger RNA vaccines compared with T-cell independent polysaccharide vaccines.27 Together these data suggest that concerted immune responses that may include the help of cytokine-producing NK cells yield the best antibody concentrations after vaccination.27,28
Almost 50% of patients did not reach S1 IgG ≥ 300 BAUs/mL. Many of these patients nevertheless demonstrated a clear increase in antibody concentrations after each vaccination, suggesting that antibody responses can be further enhanced with one or more extra vaccinations.29-31 Ideally, such vaccination schedules are guided by S1 IgG concentrations, and we are currently testing the feasibility of such an approach. Other important questions presently under investigation by us and others refer to the functionality of T and NK cells and the durability and functionality of antibody and B-cell recall responses, in particular, in patients with a reduced B-cell repertoire, patients with quantitatively or qualitatively insufficient T-cell help, and patients who are receiving daratumumab to deplete malignant plasma cells. Importantly, blunted antibody responses in rituximab-treated patients with rheumatology did not rule out the generation of T-cell immunity against SARS-CoV-2,32,33 but true vaccine efficacy in B-cell–depleted patients remains to be determined. We did observe breakthrough infections after completion of the primary vaccination schedule, but none of our study participants died of COVID-19. Although this may seem encouraging, it is important to emphasize that our study was not powered to investigate vaccine effectiveness. Indeed, an English population-based study demonstrated persistently increased risk of COVID-related death for patients with hematologic conditions despite vaccination.34 It is advised to consider SARS-CoV-2 monoclonal antibody therapy for patients with impaired humoral immunity who tested positive for SARS-CoV-2.35,36 Specific recommendations may vary depending on the dominant variant of concern in a particular region (see, for up-to-date guidelines, eg, www.covid19treatmentguidelines.nih.gov/therapies/).
Taken together, we observed that the majority of severely immunocompromised hematology patients on or early after (immuno-)chemotherapy or HCT obtained adequate antibody concentrations after 2-dose mRNA-1273 vaccination. Clinical consequences of these findings are summarized in Table 2. The most important message is that immunocompromised hematology patients should not be precluded from vaccination, even though full antibody-mediated protection cannot be guaranteed for all.
Implications of findings for COVID-19 vaccination guidelines
| Condition | Advice |
|---|---|
| Myeloid malignancies | Do not postpone vaccination during chemotherapy or therapy with TKI |
| Antibody responses are less during hypomethylating or ruxolitinib therapy | |
| Lymphoid malignancies | Do not postpone vaccination during active treatment of multiple myeloma |
| Effective B-cell–depleting therapy precludes generation of antibody responses, although B-cell numbers do not need to be normalized to generate sufficient antibody concentrations | |
| Do not interrupt B-cell–depleting therapy for vaccination as B-cell reconstitution to levels sufficient for the generation of antibody responses takes at least 8 mo | |
| Ibrutinib and venetoclax hamper potent antibody responses | |
| Autologous HCT | Multiple myeloma: vaccination is immunogenic immediately after autologous HCT |
| B-NHL: sufficient antibody responses cannot be generated<8 mo after autologous HCT | |
| Allogeneic HCT | Vaccination can be immunogenic as early as 4 mo after allogeneic HCT |
| Vaccination is immunogenic in most patients with cGVHD | |
| CAR T-cell therapy | Effective B-cell–depleting therapy precludes generation of antibody responses |
| Sickle cell disease | Vaccination is immunogenic despite functional asplenia and the use of hydroxyurea |
| Determinants of response | IgG4 concentration, B- and NK-cell numbers, number of immunosuppressants used |
| Condition | Advice |
|---|---|
| Myeloid malignancies | Do not postpone vaccination during chemotherapy or therapy with TKI |
| Antibody responses are less during hypomethylating or ruxolitinib therapy | |
| Lymphoid malignancies | Do not postpone vaccination during active treatment of multiple myeloma |
| Effective B-cell–depleting therapy precludes generation of antibody responses, although B-cell numbers do not need to be normalized to generate sufficient antibody concentrations | |
| Do not interrupt B-cell–depleting therapy for vaccination as B-cell reconstitution to levels sufficient for the generation of antibody responses takes at least 8 mo | |
| Ibrutinib and venetoclax hamper potent antibody responses | |
| Autologous HCT | Multiple myeloma: vaccination is immunogenic immediately after autologous HCT |
| B-NHL: sufficient antibody responses cannot be generated<8 mo after autologous HCT | |
| Allogeneic HCT | Vaccination can be immunogenic as early as 4 mo after allogeneic HCT |
| Vaccination is immunogenic in most patients with cGVHD | |
| CAR T-cell therapy | Effective B-cell–depleting therapy precludes generation of antibody responses |
| Sickle cell disease | Vaccination is immunogenic despite functional asplenia and the use of hydroxyurea |
| Determinants of response | IgG4 concentration, B- and NK-cell numbers, number of immunosuppressants used |
Conclusions are based on S1 IgG concentrations obtained 4 wk after the standard 2-dose mRNA-1273 vaccination schedule given 28 d apart. Other markers of immunogenicity, such as antigen-specific T-cell responses, are not taken into consideration here.
Acknowledgments
The authors extend their deepest gratitude to all patients, colleagues, students, and volunteers from participating institutes, The Netherlands Cancer Registry, and the National Institute for Public Health and the Environment, who made this study possible.
This study was financially supported by the Dutch Research Council (NWO/ZonMW, grant 10430072010009) and Amsterdam UMC.
Funding support for this article was provided by the Dutch Research Council (NWO / ZonMW) (10430072010009)
Authorship
Contribution: S.Z., A.P.K., B.I.L.-W., A.G., C.E.R., I.S.N., and M.D.H. developed the study protocol, with contributions from N.Y.R.; S.H., R.S.v.B., G.d.H., M.S.B., N.J.E.H., D.M.d.R., J.v.M., J.C., N.A.K., D.W., S.S.W., E.M.M.v.L., H.J.B., S.T.-A., E.R.-K., Q.H., K.G., L.C., L.S., Y.M.d.H., B.T., I.M.J.K., E.d.P., W.A.D., M.D.E., T.G., R.C.N.P., S.R.J., E.v.D., M.P., J.A.B., J.H.B., G.S., I.S.N., M.D.H., T.v.M., P.G.N.J.M., J.A.v.D., A.E.C.B., and C.E.R. recruited and vaccinated patients, performed experiments, and collected data; S.H., B.I.L.-W., R.S.v.B., G.d.H., M.J.v.G., I.S.N., and M.D.H. analyzed and interpreted data and wrote the manuscript; and M.H.M.H., R.W.S., Q.H., S.Z., A.P.K., T.v.M., P.G.N.J.M., J.A.v.D., A.E.C.B., A.G., and C.E.R. contributed to the manuscript and approved its final version.
Conflict-of-interest disclosure: J.C. is consultant for Novartis and received research funding from Novartis, Merus, Takeda, Genentech, and BD Biosciences and royalties from Navigate and BD Biosciences. A.P.K. is consultant for Abbvie, Janssen, Astra Zeneca, BMS, Roche, LAVA and received speakers fees from Abbvie and research funding from Janssen, Abbvie, Astra Zeneca, BMS, Celgene and Roche. S.Z. received research funding from Takeda and Janssen and participated on advisory boards of Takeda, Janssen, BMS/Celgene, Sanofi, and Oncopeptides. T.v.M. is a consultant for Janssen and Kite and received research funding and honoraria from BMS/Celgene, Genentech, and Kite. P.G.N.J.M. received research funding from Astra Zenica and is consultant for GSK and BMS. I.S.N. is consultant for Janssen, BMS/Celgene, Amgen, and Sanofi. Amsterdam UMC (M.J.v.G. and R.W.S.) filed a patent application on SARS-CoV-2 monoclonal antibodies. None of these are conflicts of interest in the present study. The remaining authors declare no competing financial interests.
Correspondence: Mette D. Hazenberg, Department of Hematology, Amsterdam University Medical Center, location AMC, Meibergdreef 9, 1105 AZ Amsterdam, The Netherlands; e-mail: m.d.hazenberg@amsterdamumc.nl.

