

Key points
We identified a novel GP1BA variant in the LRR5 domain of GPIbα (p.Arg127Gln) in a patient with a mild PT-VWD phenotype.
GOF variants in the LRR of GPIbα alter the dynamics of the C-terminal disulfide loop generating a conformation with high affinity for VWF.

Abstract
Gain-of-function (GOF) variants in GP1BA cause platelet-type von Willebrand disease (PT-VWD), a rare inherited autosomal dominant bleeding disorder characterized by enhanced platelet GPIbα to von Willebrand factor (VWF) interaction, and thrombocytopenia. To date, only 6 variants causing PT-VWD have been described, 5 in the C-terminal disulfide loop of the VWF-binding domain of GPIbα and 1 in the macroglycopeptide. GOF GP1BA variants generate a high-affinity conformation of the C-terminal disulfide loop with a consequent allosteric conformational change on another region of GPIbα, the leucine-rich-repeat (LRR) domain. We identified a novel GP1BA variant (p.Arg127Gln) affecting the LRR5 domain of GPIbα in a boy with easy bruising and laboratory test results suggestive of PT-VWD. We thus aimed to investigate the impact of the p.Arg127Gln variant on GPIbα affinity for VWF and GPIbα structure. Chinese hamster ovary cells expressing p.Arg127Gln GPIbα showed increased binding of VWF induced by ristocetin and enhanced tethering on immobilized VWF as compared with cells expressing wild-type GPIbα. Surface plasmon resonance confirmed that p.Arg127Gln enhances the binding affinity of GPIbα for VWF. Hydrogen‐deuterium exchange mass spectrometry showed that p.Arg127Gln of LRR, while having little effect on the dynamics of the LRR locally, enhances the conformational dynamics of the GPIbα C-terminal disulfide loop structure. Our data demonstrate for the first time that GOF variants outside the GPIbα C-terminal disulfide loop may be pathogenic and that aminoacidic changes in the LRR may cause allosterically conformational changes in the C-terminal disulfide loop of GPIbα, inducing a conformation with high affinity for VWF.
Introduction
Platelet-type von Willebrand disease (PT-VWD) is a rare and possibly underdiagnosed autosomal dominant bleeding disorder due to gain of function (GOF) variants in GP1BA.1 These variants confer to platelet GPIbα enhanced affinity for von Willebrand factor (VWF) and associate with a hemorrhagic diathesis of variable severity and mild thrombocytopenia with enhanced platelet volume. Thrombocytopenia is caused by a defect of proplatelet formation by megakaryocytes associated with an increased clearance of VWF-bound platelets from the circulation.2 Moreover, platelets from PT-VWD are dysfunctional, and this contributes to the bleeding phenotype.3,4
GP1BA encodes for GPIbα, the largest polypeptide in the GPIb-IX-V complex, which binds most of the extracellular ligands and cytoplasmic molecules interacting with it. The extracellular region of GPIbα is composed of an N-terminal ligand-binding domain which interacts with VWF, a polymorphic mucin-like macroglycopeptide region, and a membrane-proximal region containing a mechanosensitive domain and some cysteine residues forming disulfide bonds with GPIbβ.5,6 The ligand-binding domain is characterized by 7 leucine-rich-repeats (LRR) flanked by conserved disulfide loop structures at both the N- and C-termini.5,6 The LRR correspond to structural units, each consisting of a β strand and an α helix connected by loops. These structural units are configured in a curved shape with a parallel β-sheet on the concave side and mostly helical elements on the convex side.7
The ligand-binding domain forms a cupped-hand-like structure, with the palm of the hand representing the concave β-sheet surface of the LRR, the fingertips representing the N-terminal disulfide loop, and the opposable thumb representing the C-terminal disulfide loop.8 The C-terminal disulfide loop, known as the β-switch, is the region crucial for the binding to the A1 domain of VWF,9 and it harbors 5 of the 6 GOF variants of GPIbα causing PT-VWD described so far: p.Trp246Leu (without signal peptide: p.Trp230Leu),10 p.Gly249Val (p.Gly233Val),11 p.Gly249Ser (p.Gly233Ser),12 p.Asp251Tyr (p.Asp235Tyr),13 p.Met255Val (p.Met239Val).14 A sixth variant, p.436-444del9 (p.420-428del), is localized in the macroglycopeptide.15
Extensive characterization of the LRR region of the ligand-binding domain has identified some residues or positions that are critical for its structural integrity. Indeed, the structure of the LRR undergoes significant changes if amino acid changes involve its concave side, while the effects of variants involving its convex side were considered more difficult to predict.7,9
Recently, hydrogen‐deuterium exchange mass spectrometry (HXMS) allowed to prove that GOF variants in the C-terminal disulfide loop causing PT-VWD favor a disordered, high‐affinity conformation of the C-terminal disulfide loop, in turn causing its dissociation from the LRR of GPIbα and the creation of high‐affinity interactions with the VWF A1 domain. However, in the presence of PT-VWD variants, the disordered conformation localizes not only in the C-terminal disulfide loop but also at the convex side of the LRR, predominantly within repeats 5 and 6, which then flex in a grasping manner. This suggests an allosteric conformational linkage between the C-terminal disulfide loop and the convex side of the LRR.16
Here we report the first example of a GP1BA GOF variant in the LRR5 domain conferring to GPIbα increased affinity for VWF and generating a PT-VWD phenotype in a patient with a mild bleeding diathesis, demonstrating that GOF variants outside the C-terminal disulfide loop may be pathogenic. Moreover, we show for the first time that single amino acid changes in the LRR domain may cause conformational changes in the C-terminal disulfide loop of GPIbα, inducing high affinity for VWF.
Methods
Clinical case
A 14-year-old boy was referred to our center for a mild bleeding diathesis. Since his first years of life, he suffered easy bruising, recurrent epistaxis which required cauterization, and occasional short-lasting gum bleeding. The ISTH-BAT bleeding score was 4, which is just above normal for a male subject.17 He was an only child, and his mother did not refer any bleeding symptoms. The father was not available for collection of clinical history or platelet function testing.
The patient was studied on 4 different occasions over a 3-year period, and 4 healthy controls were studied in parallel. The proband’s mother and controls gave their informed, written consent to the studies performed. All human studies were approved by the responsible Institutional review boards (CEAS Umbria, approval n. 2663/15), and all studies were carried out in conformity with the Declaration of Helsinki.
Routine blood clotting assays, including VWF:Ag, VWF:RCo, and VWF:CBA, were normal. Bleeding time, as assessed by the Mielke method,18 was normal (4.5 minutes; controls: 5.5 ± 2 minutes, min-max 2.5-10.5) as well as the PFA-100 (Dade-Behring, Deerfield, IL) closure time performed using collagen/ADP (C/ADP) and collagen/epinephrine (C/Epi) cartridges.19 Platelet count was normal (208x106 platelets/mL, min-max 180-215; controls: 237 ± 35 × 106 platelets/mL, min-max 174-289) with increased mean platelet volume (MPV) (12 fL; controls: 8.3 ± 0.6 fL, min-max 7.2-9.4). Platelet aggregation in response to collagen, ADP, arachidonic acid, and adrenaline was normal while ristocetin-induced platelet agglutination (RIPA) was increased (0.6-0.7 mg/mL; normal values 0.85 ± 0.1 mg/mL, range 0.80-1.05 mg/mL). VWF binding to platelets assessed by flow cytometry after the addition of 0.7 mg/mL of ristocetin in platelet-rich plasma (PRP) was increased (mean fluorescence intensity 1.49, normal values 0.58-0.98). Platelet agglutination induced by cryoprecipitate (60 U/dL of VWF)20,21 was absent. All the main platelet glycoproteins, including GPIbα, assessed by flow cytometry,22 were normal. Therefore, after exclusion of VWD and of other inherited platelet function disorders according to published guidelines,23,24 and given increased RIPA, we decided to perform laboratory tests for PT-VWD diagnosis.23
Results of all the diagnostic laboratory assays are shown in supplemental Table 1.
RIPA mixing test
RIPA was carried out in citrated PRP using an optical aggregometer (APACT-4, Helena Biosciences Europe, Sunderland, UK) as reported.19,20 Aggregometric mixing assays were performed by adding patient or control platelet-poor plasma to patient or control platelets in the following combinations: patient platelets/patient plasma, patient platelets/control plasma, control platelets/control plasma, control platelets/patient plasma.20
Platelet VWF-binding by flow cytometry
The binding of VWF to platelets induced by ristocetin was evaluated in PRP by flow cytometry using a mouse anti-human VWF antibody, clone 4f9 (Immunotech, Marseille, France) and a FITC-conjugated goat anti-mouse IgG (Beckman Coulter, Miami, FL), as previously described.19,20 Flow-cytometric mixing assays were performed by adding patient or control platelet-poor plasma to patient or control platelets in the combinations reported above for the RIPA mixing test. Samples were analyzed in a CytoFLEX flow cytometer (Beckman Coulter, Miami, FL).20
Sanger sequencing of the GP1BA gene
Genomic DNA was isolated from patient peripheral blood, his mother, and 50 healthy controls. The entire coding region of the GP1BA gene was amplified by PCR in 4 overlapping fragments using a series of oligonucleotide primer pairs (primer sequences are provided in supplemental Table 2). Upon identification of the GP1BA variant in the patient, PCR was performed with the DNA of the mother and controls using primers flanking only the region of interest. PCR products were purified using the Wizard SV Gel and PCR Clean-Up System kit (Promega, Milan, Italy). Sequencing was performed in an AB3500 genetic analyzer (Applied Biosystems, Monza, Italy).25
Generation of the mutant GP1BA plasmids
The pDX- GPIbαWT plasmid, coding for human GPIbα, was kindly given by J.A. Lopez (Puget Sound Blood Center, Seattle, WA) and was used to generate mutants. GPIbα mutants p.Gly249Val and p.Arg127Gln were generated by site-directed mutagenesis as described previously.26,27 Primer sequences used to generate GPIbα mutants are reported in supplemental Table 2. Successful mutagenesis was verified by Sanger sequencing.
CHO β/IX cells transfection
Chinese hamster ovary (CHO) β/IX cells, stably expressing the GPIbβ and GPIX subunits of the receptor complex,28 were generated by F. Lanza (UMR S_949 - EFS-Alsace, Strasbourg, France). Cells were grown in DMEM-F12 medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 1% l-glutamine. To maintain high expression of the GPIbβ and GPIX polypeptides, the medium was periodically supplemented with methotrexate (MTX) 80 mmol/L and geneticin 400 μg/mL (Invitrogen, Life Technologies, Monza, Italy). Cells were incubated at 37°C in 5% CO2. Plasmids were transfected transiently into CHO β/IX cells using the Turbofect in vitro transfection reagent (Fermentas, Life Technologies). As negative control, CHO β/IX cells were transfected with the empty vector.
GPIbα expression by CHO β/IX cells
Forty-eight hours after transfection, CHO cells were detached from culture plates, washed, and resuspended in phosphate-buffered saline (PBS) at a concentration of 106/mL. Fifty μl of cells were incubated with 0.5 μg of monoclonal antibody LJ-P19, recognizing GPIbα29 (L. De Marco, Department of Translational Research, National Cancer Center, Aviano, Italy) and a FITC-conjugated goat anti-mouse IgG (0.01 mg/mL, Beckman Coulter) for 30 minutes at room temperature and analyzed by flow cytometry.30
Binding of VWF to CHO cells induced by ristocetin
Fifty μl of transfected CHO β/IX cells resuspended at a concentration of 106/mL were incubated with 8 μg/mL of purified human VWF31 (L. De Marco, Department of Translational Research, National Cancer Center, Aviano, Italy) and different concentrations of ristocetin (from 0 to 2.5 mg/mL) for 5 minutes at 37°C. Fifty μl of PFA 2% were then added to the mixture, and VWF-binding was assessed by flow cytometry as described above.
Real-time microscopy perfusion studies
Rolling of transfected CHO β/IX cells on a VWF-coated surface under flow conditions was assessed in a laminar-flow perfusion chamber.32 Glass coverslips coated with 100 μg/mL of purified human VWF overnight at 4°C and then blocked with 1% human serum albumin in PBS for 90 minutes at room temperature were placed in the chamber. Real-time perfusion studies were carried out using CHO cells resuspended at a concentration of 106/mL in DMEM-F12 medium. The cell suspension was perfused for 10 minutes at the shear rate of 250 seconds−1. Cell rolling was continuously recorded by an inverted microscope (Zeiss, ObserverZ.1, Carl Zeiss, Jena, Germany) equipped with AxioCam MRm (Carl Zeiss).32 Images were captured at 1-second intervals for the 10-minute perfusion period and then analyzed offline with ImageJ v. 1.52 TrackMate (v5.2.0) software.33
Expression and purification of p.Arg127Gln for Spectroscopy, SPR, DSC, and HXMS
Wild-type (WT) and p.Arg127Gln GPIbα (amino acids Met1-Glu301) were expressed in the pIRESneo2 plasmid vector in HEK293 cells as fusion construct containing a C-terminal Hexahistidine-tag and a C-terminal Flag-tag. Both proteins were purified via Ni-NTA Sepharose, and their quality was confirmed via RP-HPLC and with UV-Vis, fluorescence, and circular dichroism spectra (supplemental Figure 1; supplemental Methods 2.1 and 2.2).
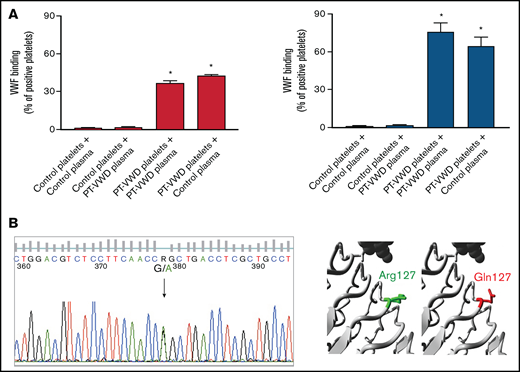
A novel GP1BA variant in a PT-VWD patient. (A) Platelet/plasma mixing studies for VWF binding to platelets induced by ristocetin (1 mg/mL) assessed by flow cytometry. VWF binding was measured in the following mixtures: control platelets/control plasma, control platelets/proband (or PT-VWD) plasma, proband (or PT-VWD) platelets/proband (or PT/VWD) plasma and proband (or PT-VWD) platelets/control plasma. Binding of VWF to platelets is expressed as percent of positive platelets. Data are means ±SEM from 4 independent experiments, * = P < .05 vs control platelets + control plasma; Two-way ANOVA. (B) Sequencing of DNA from the proband, showing the heterozygous c.G380A variant in GP1BA (NM_000173.7) leading to p.Arg127Gln. On the right, close-up of the variant obtained by HOPE. GPIbα is colored gray, the side chains of both the WT and the mutant residue are shown and colored green and red, respectively.
A novel GP1BA variant in a PT-VWD patient. (A) Platelet/plasma mixing studies for VWF binding to platelets induced by ristocetin (1 mg/mL) assessed by flow cytometry. VWF binding was measured in the following mixtures: control platelets/control plasma, control platelets/proband (or PT-VWD) plasma, proband (or PT-VWD) platelets/proband (or PT/VWD) plasma and proband (or PT-VWD) platelets/control plasma. Binding of VWF to platelets is expressed as percent of positive platelets. Data are means ±SEM from 4 independent experiments, * = P < .05 vs control platelets + control plasma; Two-way ANOVA. (B) Sequencing of DNA from the proband, showing the heterozygous c.G380A variant in GP1BA (NM_000173.7) leading to p.Arg127Gln. On the right, close-up of the variant obtained by HOPE. GPIbα is colored gray, the side chains of both the WT and the mutant residue are shown and colored green and red, respectively.
The VWF A1 domain required for the surface plasmon resonance (SPR) experiments was refolded from inclusion bodies and purified as previously described.34,35 All proteins were dialyzed into PBS (10 mM NaPhosphate, 150 mM NaCl, PH 7.4) and stored on ice for a maximum of 2 weeks. Prior to any experiment, protein solutions were centrifuged for 10 minutes at 60 000 g and 4°C to remove any aggregates.
SPR
SPR experiments were performed on a Biacore T100 using a CM5 chip at 25°C as previously described.16,36 Anti-Flag M2 Antibody (Sigma Aldrich) was diluted in 10 mM NaAcetate (pH 4.5) to a concentration of 50 µg/mL and immobilized to a level of ∼10 000 RU on all 4 channels of the CM5 chip using an amine coupling kit (Cytiva Life Sciences).
Interactions between VWF A1 and the anti-Flag captured GPIbα were measured in HBS-EP buffer (Cytiva Life Sciences) at a flow rate of 30 µL/min. Each binding cycle began with loading 3 µM WT or p.Arg127Gln GPIbα, followed by a 100-second dissociation phase. Then VWF A1 was allowed to bind for 100 seconds, followed by a 200-second dissociation phase. Between cycles, the chip surface was regenerated with 10 mM glycine pH 2.0. Binding responses were referenced and blank subtracted prior to analysis to remove nonspecific interactions and baseline drifts. A1-response levels were normalized by the GPIbα-capture level to account for differences in the GPIbα binding. Equilibrium responses were plotted as a function of A1 concentration and globally fitted to a solution affinity model in Sigmaplot 12.5 as previously described.16
Differential scanning calorimetry
Differential scanning calorimetry (DSC) of WT and p.Arg127Gln GPIbα was performed in PBS buffer at scan rates of 2.0, 1.5, 1.0, 0.75, and 0.5°C/minute on a TA Instruments NanoDSC at a constant pressure of 3 atm. Prior to the measurement, all protein and buffer solutions were degassed under moderate stirring. The instrument was equilibrated with scans between 10 and 95°C with PBS loaded into the sample and reference capillaries. Then the protein solution was loaded into the sample capillary. All scans were processed using the NanoAnalyze Software (v 3.11.0) provided with the instrument. The DSC thermogram was background corrected by subtracting the following irreversible scan as baseline, and the analysis using a two-state irreversible model was performed as previously described.34
HXMS
HXMS as a function of incubation time was performed on WT and p.Arg127Gln GPIbα using an LTQ Orbitrap XL mass spectrometer. Samples containing 80% D2O were prepared by 1 to 5 dilution of the proteins into Tris-buffered saline in D2O (pD 7.4). After incubating the sample between 1 minute to overnight at 25°C, the exchange was stopped by a drop in pD to ∼2.3 using a concentrated HCl solution. Quenched samples were then loaded and digested on a custom-packed pepsin column under isocratic flow (200 µL/minute) of 0.5% (vol/vol) formic acid using a modified Agilent 1100 G2226 Nano Pump. Proteolytic peptides were captured by a custom-packed C8 trap column. After 5 minutes, the trap was switched inline with a water-acetonitrile gradient provided by a Waters nanoAcquity UPLC gradient pump. The solvent flow was 15 µL/min. Water with 0.5% formic acid was used as solvent A and 80% Acetonitrile adjusted to a pH of 2.3 was used as solvent B. Peptides were eluted off the trap column and separated on a subsequent C18 column followed by the injection into the mass spectrometer.
Prior to the exchange experiments, peptide maps with a library of searchable peptides were generated from multiple all H experiments in absence of deuterium (supplemental Figure 2). These maps were generated using Bioworks 3.3.1 (Thermo Fisher Scientific) and EXMS2 as previously described.16,36 Deuterated peptides were identified with EXMS2 using the generated peptide library and the first all H experiment as reference. The subsequent HDsite analysis37 was performed using a temperature of 25°C, a pD of 7.4, and a deuteration range of 0.8. After the analysis, switchable peptides were averaged manually. A table with experimental parameters is provided in the supporting information (supplemental Table 3).
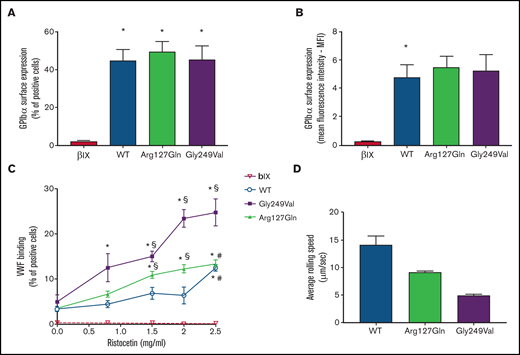
The p.Arg127Gln variant increases the binding of VWF to GPIbα. (A) and (B) Surface expression of GPIbα in CHO β/IX cells transfected with WT, p.Arg127Gln or p.Gly249Val as assessed by flow cytometry using the LJ-P19 antibody and a FITC-conjugated goat anti-mouse IgG shown as percentage of positive cells (A) and mean fluorescence intensity (MFI) (B) (n = 10, * P < .05 vs CHO β/IX, one-way ANOVA). (C) Binding of VWF to CHO β/IX cells transfected with WT, p.Arg127Gln, or p.Gly249Val as assessed by flow cytometry using a mouse anti-human VWF antibody, clone 4f9, and a FITC-conjugated goat anti-mouse IgG after incubation with 8 μg/ml of purified human VWF and different concentrations of ristocetin for 5 minutes at 37°C (n = 10, * P < .05 vs 0 mg/ml; § P < .05 vs WT; # P < .05 vs Gly249Val; two-way ANOVA). (D) Average rolling speed of CHO β/IX cells transfected with WT, p.Arg127Gln, or p.Gly249Val on a VWF-coated surface under flow conditions assessed in a laminar-flow perfusion chamber. Cell rolling was continuously recorded using a Zeiss, ObserverZ.1 (Carl Zeiss, Jena, Germany) inverted microscope equipped with AxioCam MRm (Carl Zeiss). Images were captured at 1-second intervals for 10 minutes and then analyzed offline with the ImageJ v. 1.52 TrackMate (v5.2.0) software (n = 4, * P < .05 vs 0mg/ml; § P < .05 vs WT, # P < .05 vs p.Gly249Val, one-way ANOVA). CHO β/IX cells transfected with the empty vector did not tether to the VWF-coated surface and flowed away without rolling, as shown in supplemental Movie 4.
The p.Arg127Gln variant increases the binding of VWF to GPIbα. (A) and (B) Surface expression of GPIbα in CHO β/IX cells transfected with WT, p.Arg127Gln or p.Gly249Val as assessed by flow cytometry using the LJ-P19 antibody and a FITC-conjugated goat anti-mouse IgG shown as percentage of positive cells (A) and mean fluorescence intensity (MFI) (B) (n = 10, * P < .05 vs CHO β/IX, one-way ANOVA). (C) Binding of VWF to CHO β/IX cells transfected with WT, p.Arg127Gln, or p.Gly249Val as assessed by flow cytometry using a mouse anti-human VWF antibody, clone 4f9, and a FITC-conjugated goat anti-mouse IgG after incubation with 8 μg/ml of purified human VWF and different concentrations of ristocetin for 5 minutes at 37°C (n = 10, * P < .05 vs 0 mg/ml; § P < .05 vs WT; # P < .05 vs Gly249Val; two-way ANOVA). (D) Average rolling speed of CHO β/IX cells transfected with WT, p.Arg127Gln, or p.Gly249Val on a VWF-coated surface under flow conditions assessed in a laminar-flow perfusion chamber. Cell rolling was continuously recorded using a Zeiss, ObserverZ.1 (Carl Zeiss, Jena, Germany) inverted microscope equipped with AxioCam MRm (Carl Zeiss). Images were captured at 1-second intervals for 10 minutes and then analyzed offline with the ImageJ v. 1.52 TrackMate (v5.2.0) software (n = 4, * P < .05 vs 0mg/ml; § P < .05 vs WT, # P < .05 vs p.Gly249Val, one-way ANOVA). CHO β/IX cells transfected with the empty vector did not tether to the VWF-coated surface and flowed away without rolling, as shown in supplemental Movie 4.
Results
Binding of VWF to patient’s platelets is increased
Mixing tests performed by either light transmission aggregometry (supplemental Table 1) or flow cytometry showed that patient’s plasma did not affect reactivity of control platelets to ristocetin while patient’s platelets maintained enhanced VWF-binding also when mixed with control plasma, demonstrating the platelet origin of the defect. The degree of enhancement of VWF-binding was smaller than that of a PT-VWD patient carrying the known pathogenic variant p.Met239Val GP1BA20 (Figure 1A).
Identification of a novel GP1BA variant in the LRR5
DNA sequencing revealed a heterozygous c.G380A variant in GP1BA (NM_000173.7) (Figure 1B), resulting in a missense substitution of an arginine with a glutamine at position 127 (p.Arg127Gln with signal peptide, p.Arg111Gln without signal peptide). No other variants were detected in the GP1BA gene. The variant was absent from the mother and 50 healthy controls (100 alleles) but is present in the gnomAD database (rs749454966), but only in 8 alleles, thus with a very low frequency (exomes allele frequency = 0.0000201; genomes allele frequency = 0.0000956; total allele frequency = 0.00002851).38,39 In silico analysis of pathogenicity with 4 different prediction softwares classified the variant as benign (supplemental Table 4). Moreover, Arg127 is not a highly conserved residue (supplemental Table 5). Therefore, according to the ACMG guidelines,40 we provisionally classified the variant as of uncertain significance (VUS) (supplemental Table 6). The variant is in the LRR5 domain, 1 of the 7 LRR of GPIbα. In silico protein modeling carried out using HOPE41 predicted that the positive charge of arginine is lost after the substitution with glutamine. Moreover, glutamine is smaller than arginine, and these characteristics might modify the structure of the protein and/or its interactions with other proteins (Figure 1B).
To exclude the presence of pathogenic variants in other genes causative of platelet disorders, DNA of the proband was analyzed with a targeted sequencing platform able to analyze 72 genes associated with inherited platelet disorders or significant in platelet physiology.42 Analysis with DIGEVAR (Discovering Genetic Variants), a web tool developed in-house for user-friendly analysis of HTS data (https://digevar.imib.es), confirmed that only this rare variant in GP1BA was compatible with the patient phenotype (supplemental Table 7).
The p.Arg127Gln variant increases the binding of VWF to GPIbα
The p.Arg127Gln variant did not affect the synthesis or surface expression of GPIbα, as shown by flow cytometric analysis highlighting normal expression of the receptor on transfected CHO cells (Figure 2A, B), as already observed in patient platelets (supplemental Table 1).
Therefore, in order to determine whether the new variant on GPIbα modulated its affinity for VWF, we analyzed the binding of VWF to CHO cells expressing WT GPIbα, p.Arg127Gln GPIbα, and p.Gly249Val GPIbα, a known PT-VWD GOF variant involving the C-terminal disulfide loop,11 upon incubation with ristocetin. CHO cells expressing WT GPIbα bound VWF only in the presence of the highest dose of ristocetin (2.5 mg/mL) while the lowest dose of ristocetin triggering the binding of VWF to CHO cells expressing the well-known PT-VWD variant p.Gly249Val GPIbα was 0.8 mg/mL, confirming the enhanced affinity of p.Gly249Val GPIbα for VWF. CHO cells expressing the novel p.Arg127Gln GPIbα variant bound VWF in the presence of 1.5 mg/mL of ristocetin, showing that the affinity of p.Arg127Gln GPIbα for VWF is increased with respect to WT cells but less compared with p.Gly249Val GPIbα (Figure 2C).
The p.Arg127Gln variant induces increased tethering of GPIbα to VWF under flow conditions
CHO β/IX cells expressing WT, p.Arg127Gln, and p.Gly249Val GPIbα did roll on immobilized VWF, with cells expressing the WT form rolling significantly faster. The rolling speed of CHO cells expressing the novel p.Arg127Gln GPIbα variant was significantly lower than that of WT cells, although still higher than the rolling speed of CHO cells expressing the well-known p.Gly249Val GPIbα (Figure 2D, supplemental Movies 1-3). CHO β/IX cells transfected with the empty vector did not tether to the VWF-coated surface and flowed away without rolling (supplemental Movie 4).
The affinity of p.Arg127Gln GPIbα for VWF is enhanced and associated with reduced thermodynamic stability
The affinity of p.Arg127Gln GPIbα for A1, assessed by SPR, was slightly increased as shown by the equilibrium binding curve for p.Arg127Gln, which is shifted toward a lower ligand concentration in comparison with WT GPIbα, although lesser than the affinity of other PT-VWD mutants that were described previously16 (Figure 3A). The equilibrium association constant, KD, of p.Arg127Gln GPIbα was significantly decreased compared with the WT GPIbα (2.04 ± 0.07 μM vs 3.5 ± 0.1 μM), although less if compared with other typical PT-VWD mutant GPIbα proteins (p.Trp246Leu: 0.06 ± 0.002 μM; p.Gly249Val: 0.23 ± 0.006 μM; p.Met255Val: 0.36 ± 0.002 μM).16
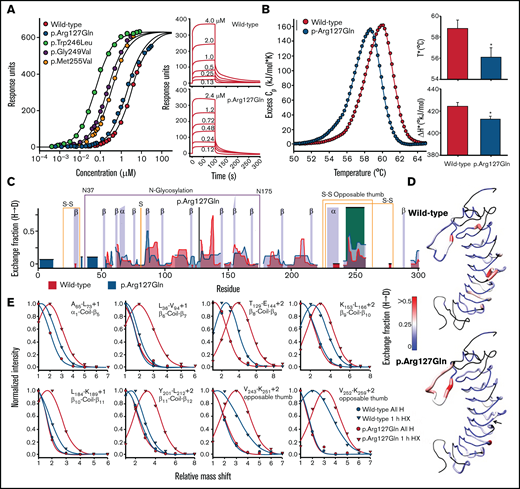
p.Arg127Gln allosterically enhances the conformational dynamics of the C-terminal disulfide loop of GPIbα. (A) Equilibrium binding of VWF A1 to WT and p.Arg127Gln GPIbα obtained from the maximum SPR response as a function of A1 concentration. Data for p.Trp246Leu, p.Gly249Val and p.Met255Val were taken from Tischer et al 2019.16 Maximum SPR response (Rmax) is 639 ± 4 response units. Panels on the right show representative SPR sensorgrams as a function of A1 concentration. The SPR data indicate that p.Arg127Gln is a mild GOF mutation of GPIbα, as its affinity is slightly enhanced relative to WT. (B) Excess heat capacity curves for thermal unfolding of WT and p.Arg127Gln GPIbα obtained by DSC at a scan rate of 2°C/min. Thermal unfolding is irreversible and dependent on the thermal scan rate. Slower thermal scan rates are shown in the supporting information (supplemental Figure 3). Fitting parameters are reported in supplemental Table 3 . Panels on the right show the scan rate independent thermal transition temperatures (T* in °C) and enthalpies (ΔH* in kJ/mol) where the rate of unfolding, kunf = 1. Asterisks (*) on the p.Arg127Gln bars indicate a P < 0.05 relative to WT. DSC demonstrates that p.Arg127Gln destabilizes GPIbα, as evident by a diminished T* and ΔH* relative to WT GPIbα. (C) Hydrogen-deuterium Exchange fraction of WT (blue) and p.Arg127Gln GPIbα (gray) as a function of residue number after 1 hour of incubation at 25°C in 80% (vol/vol) deuterium oxide. Additional timepoints ranging 1 minute to 18 hours overnight incubation are included in supplemental Figure 4. Parameters for the exchange experiments are summarized in supplemental Table 8. The opposable thumb region of GPIbα is highlighted in orange. The position of the p.Arg127Gln mutation is indicated by a black vertical line. (D) Exchange fraction mapped onto the crystal structure of GPIbα (pdb ID = 1GWB).48 Colors are as follows: black = not resolved, blue = 0, white = 0.25, and red ≥0.5. Structures were rendered using UCSF chimera.49 The arrow in p.Arg127Gln indicates the position of the mutation. (E) Peptide envelopes (normalized intensity as a function of the deuterium-induced mass shift relative to all H peaks in absence of deuterium) of 8 peptides spanning the protein after 1 hour of exchange indicate an increased deuterium uptake for p.Arg127Gln relative to WT. Envelopes represent HXMS raw data and were analyzed with EXMS237 prior to deconvolution of the HX fraction with HDsite. The peptides shown in Figure 3E represent a small portion of the HX raw data that were extracted from the MS data using EXMS2.37 A multitude of such peptide envelopes was used for the deconvoluted exchange fraction (Figure 3C,D). The HXMS data shown in panels (C)-(E) agree with the stability data shown in (B) as p.Arg127Gln destabilizes GPIbα. The primary impact of the mutation, however, can be observed in the opposable thumb region where the exchange is significantly enhanced. This indicates that p.Arg127Gln increases the conformational dynamics of this region. The difference between p.Arg127Gln and other platelet-type VWD mutations occurring within the opposable thumb sequence16 is that p.Arg127Gln acts allosterically through the LRR of GPIbα.
p.Arg127Gln allosterically enhances the conformational dynamics of the C-terminal disulfide loop of GPIbα. (A) Equilibrium binding of VWF A1 to WT and p.Arg127Gln GPIbα obtained from the maximum SPR response as a function of A1 concentration. Data for p.Trp246Leu, p.Gly249Val and p.Met255Val were taken from Tischer et al 2019.16 Maximum SPR response (Rmax) is 639 ± 4 response units. Panels on the right show representative SPR sensorgrams as a function of A1 concentration. The SPR data indicate that p.Arg127Gln is a mild GOF mutation of GPIbα, as its affinity is slightly enhanced relative to WT. (B) Excess heat capacity curves for thermal unfolding of WT and p.Arg127Gln GPIbα obtained by DSC at a scan rate of 2°C/min. Thermal unfolding is irreversible and dependent on the thermal scan rate. Slower thermal scan rates are shown in the supporting information (supplemental Figure 3). Fitting parameters are reported in supplemental Table 3 . Panels on the right show the scan rate independent thermal transition temperatures (T* in °C) and enthalpies (ΔH* in kJ/mol) where the rate of unfolding, kunf = 1. Asterisks (*) on the p.Arg127Gln bars indicate a P < 0.05 relative to WT. DSC demonstrates that p.Arg127Gln destabilizes GPIbα, as evident by a diminished T* and ΔH* relative to WT GPIbα. (C) Hydrogen-deuterium Exchange fraction of WT (blue) and p.Arg127Gln GPIbα (gray) as a function of residue number after 1 hour of incubation at 25°C in 80% (vol/vol) deuterium oxide. Additional timepoints ranging 1 minute to 18 hours overnight incubation are included in supplemental Figure 4. Parameters for the exchange experiments are summarized in supplemental Table 8. The opposable thumb region of GPIbα is highlighted in orange. The position of the p.Arg127Gln mutation is indicated by a black vertical line. (D) Exchange fraction mapped onto the crystal structure of GPIbα (pdb ID = 1GWB).48 Colors are as follows: black = not resolved, blue = 0, white = 0.25, and red ≥0.5. Structures were rendered using UCSF chimera.49 The arrow in p.Arg127Gln indicates the position of the mutation. (E) Peptide envelopes (normalized intensity as a function of the deuterium-induced mass shift relative to all H peaks in absence of deuterium) of 8 peptides spanning the protein after 1 hour of exchange indicate an increased deuterium uptake for p.Arg127Gln relative to WT. Envelopes represent HXMS raw data and were analyzed with EXMS237 prior to deconvolution of the HX fraction with HDsite. The peptides shown in Figure 3E represent a small portion of the HX raw data that were extracted from the MS data using EXMS2.37 A multitude of such peptide envelopes was used for the deconvoluted exchange fraction (Figure 3C,D). The HXMS data shown in panels (C)-(E) agree with the stability data shown in (B) as p.Arg127Gln destabilizes GPIbα. The primary impact of the mutation, however, can be observed in the opposable thumb region where the exchange is significantly enhanced. This indicates that p.Arg127Gln increases the conformational dynamics of this region. The difference between p.Arg127Gln and other platelet-type VWD mutations occurring within the opposable thumb sequence16 is that p.Arg127Gln acts allosterically through the LRR of GPIbα.
Thermal unfolding of p.Arg127Gln, determined by DSC, is irreversible and dependent on the applied thermal scan rate. The excess heat capacity curves shown in the left panel of Figure 3B compare the p.Arg127Gln with WT GPIbα and demonstrate that the mutation destabilizes GPIbα. TM,app (ie, the thermal transition temperature) and ΔHapp (ie, the area under the thermal transition) are both reduced relative to WT GPIbα. A two-state irreversible analysis of these heat capacity curves16,34 yields scan-rate independent T* and ΔH* that are significantly (P < .05) reduced for p.Arg127Gln relative to WT GPIbα (Figure 3B).
p.Arg127Gln allosterically enhances the conformational dynamics of the C-terminal disulfide loop of GPIbα
HXMS provides a means to identify local regions of protein flexibility by determining the exchange of amide backbone hydrogen atoms with deuterated water (D2O) provided in the solvent. Overall, p.Arg127Gln exhibits an exchange pattern quite similar to that of WT GPIbα, except for the C-terminal disulfide loop region (Glu241-Val259), where the HD exchange of p.Arg127Gln is enhanced relative to the WT protein, indicating an increase in conformational flexibility of this region (Figure 3C,D). Figure 3E shows the effect of the mutation on the deuterium incorporation for various peptides throughout the protein. Relative to the reference “all H” peak (determined in absence of deuterium) and the 1-hour exchange peaks for WT GPIbα, p.Arg127Gln exhibits an increased deuterium incorporation in all peptides, and thus enhanced conformational flexibility, as is evident by the shift toward higher mass.
The binding affinity of p.Arg127Gln is proportional to its thermal stability
We assessed correlations between the thermodynamic stability (thermal transition temperature, T*), the binding affinity (dissociation constant, KD), and the mean deuterium exchange fraction of the C-terminal disulfide loop. The KD of p.Arg127Gln is proportional to the T*, and this follows the trend reported for other platelet-type VWD mutations located within the C-terminal disulfide loop16 (Figure 4A). When we correlated the mean exchange fraction for the C-terminal disulfide loop with KD and T*, we observed that while there is a relation between these metrics for PT-VWD mutations in the opposable thumb sequence, p.Arg127Gln does not follow the trend as the mean exchange fraction of the C-terminal disulfide loop is higher than expected, confirming the allosteric linkage of p.Arg127Gln between the LRR and the C-terminal disulfide loop (Figure 4B).
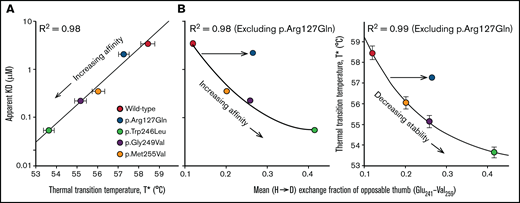
Correlations between binding affinity, thermal stability and local disorder in the opposable thumb region. Panel (A) shows the SPR binding affinity as a function of the thermal transition temperature (T*). Panel (B) correlates the binding affinity (left panel) and T* (right panel) with the average HD exchange fraction (after 1 hour) of the opposable thumb region. While p.Arg127Gln follows the trend in panel (A), it does not in panel (B). This observation demonstrates that p.Arg127Gln enhances the conformational dynamics of the opposable thumb indirectly through allostery while having little effect on the dynamics of local sequence environment near the mutation or the LRR domain.
Correlations between binding affinity, thermal stability and local disorder in the opposable thumb region. Panel (A) shows the SPR binding affinity as a function of the thermal transition temperature (T*). Panel (B) correlates the binding affinity (left panel) and T* (right panel) with the average HD exchange fraction (after 1 hour) of the opposable thumb region. While p.Arg127Gln follows the trend in panel (A), it does not in panel (B). This observation demonstrates that p.Arg127Gln enhances the conformational dynamics of the opposable thumb indirectly through allostery while having little effect on the dynamics of local sequence environment near the mutation or the LRR domain.
Discussion
In this study, starting from the identification of a novel GPIbα p.Arg127Gln variant in LRR5 in a patient with a mild PT-VWD clinical and laboratory phenotype, we show for the first time that amino acid substitutions in the LRR may cause conformational changes in the C-terminal disulfide loop of GPIbα, enhancing its affinity for VWF.
Our patient had a mild bleeding history, normal platelet number, but increased mean platelet volume and an increased RIPA, although less than typical PT-VWD, and enhanced binding of VWF to platelets as assessed by flow cytometry. GP1BA sequencing revealed the p.Arg127Gln variant, which we initially classified as a VUS, but after functional and HXMS studies we reclassified as pathogenic.
Excluding 1 family with a 27 bp deletion in the macroglycopeptide region of GPIbα,15 that is outside the ligand-binding site, all the previously reported PT-VWD variants were single nucleotide changes affecting the C-terminal disulfide loop of the ligand-binding site.10-14 The p.Arg127Gln variant identified here is the first found in the LRR5 of the VWF-binding domain.
It has been suggested that PT-VWD pathogenic variants in the ligand-binding domain induce a conformational change of the C-terminal disulfide loop from a coil to a β‐hairpin, in this way enhancing the affinity of GPIbα for VWF.43,44 However, a recent study disproved this theory, showing instead that they favor a disordered protein structure locally in the C-terminal disulfide loop (the “opposable thumb”) and allosterically in the convex side of the LRR (the “dorsum”), in particular within repeats 5 and 6. This lends to a GPIbα high‐affinity binding conformation and, like a hand, allows the extension of the opposable thumb from the palm, enabling the hand to grasp toward a fist.16
In our case, the p.Arg127Gln variant localizes exactly in the middle of the “dorsum” of GPIbα, (ie, the region affected allosterically by local changes in the C-terminal disulfide loop). Therefore, we hypothesized that, reciprocally, local changes in the GPIbα dorsum might in turn allosterically influence the structure of the thumb and, consequently, confer to GPIbα a high‐binding affinity conformation (Figure 5).
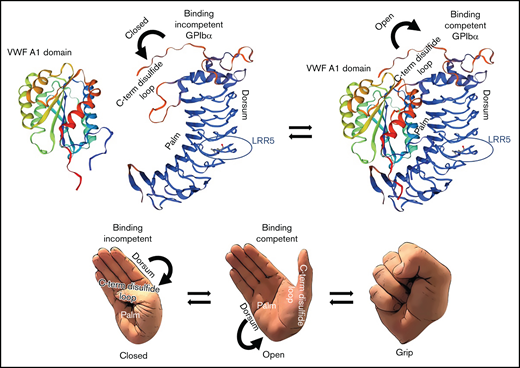
Proposed mechanism of interaction between GPIbα and the A1 domain of VWF. Proposed mechanism of interaction between GPIbα and the A1 domain of VWF based on the data from Tischer et al16 and the results of the current manuscript. GPIbα is mostly binding-incompetent when the C-terminal disulfide loop is structured and “attached” to its palm. Destabilization of the C-terminal disulfide loop enables a binding-competent conformation in which GPIbα can interact tightly with the A1 domain of VWF. Protein structure homology modeling of the ligand-binding region of GPIbα and for the A1 domain of VWF was performed using the SWISS-MODEL workspace. The SWISS-MODEL template library (GPIbα SMTL ID:3P72, PDB release 2017-01-26; VWFA1 SMTL ID: 1AUQ, PDB release 1998-10-14) was searched with Blast and HHBlits for evolutionary related structures matching the target sequence.
Proposed mechanism of interaction between GPIbα and the A1 domain of VWF. Proposed mechanism of interaction between GPIbα and the A1 domain of VWF based on the data from Tischer et al16 and the results of the current manuscript. GPIbα is mostly binding-incompetent when the C-terminal disulfide loop is structured and “attached” to its palm. Destabilization of the C-terminal disulfide loop enables a binding-competent conformation in which GPIbα can interact tightly with the A1 domain of VWF. Protein structure homology modeling of the ligand-binding region of GPIbα and for the A1 domain of VWF was performed using the SWISS-MODEL workspace. The SWISS-MODEL template library (GPIbα SMTL ID:3P72, PDB release 2017-01-26; VWFA1 SMTL ID: 1AUQ, PDB release 1998-10-14) was searched with Blast and HHBlits for evolutionary related structures matching the target sequence.
Our HXMS studies indeed demonstrate that p.Arg127Gln, while having little effect on the intrinsic dynamics of the LRR region, enhances the conformational dynamics of the opposable thumb. This confirms the existence of a bidirectional allosteric conformational linkage between the C-terminal disulfide loop and the convex side of the LRR.
This indirect effect may be the reason for the lower binding affinity for VWF of this variant compared with “classical” PT-VWD variants occurring in the opposable thumb sequence and, consequently, of the mild PT-VWD clinical and laboratory phenotype of our patient. Indeed, functional studies carried out with CHO cells and SPR studies confirmed that the binding affinity of p.Arg127Gln GPIbα to the VWF A1 domain was increased relative to that of WT GPIbα, out that PT-VWD variants affecting the C-terminal disulfide loop conferred the highest affinity.
In support of our data, and thus of the role of the LRR in the acquisition of a high-affinity conformation by GPIbα, previous in vitro functional studies demonstrated that the substitution of His86 in LRR4 with either Ala or Glu resulted in a GOF phenotype.45
The bleeding and laboratory phenotype of our patient is rather mild; therefore, it could be speculated that while an increase of the VWF-binding affinity of about 2 times would cause a mild phenotype (ISTH BAT BS: 4), a higher increase of affinity (from 9.7 for the p.Met255Val variant to 58 times for the p.Trp246Leu variant) generated by typical PT-VWD GP1BA variants would generate a more severe one (ISTH BAT BS up to 17). However, the only study that systematically assessed the bleeding phenotype of 13 PT-VWD patients carrying 6 different GP1BA variants showed that the bleeding score was variable between patients with the same mutation,46 in line with the typically variable phenotype of these patients, which worsens in stressing conditions.47 On the other hand, the VWF-binding affinity contributes to regulating platelet count because more platelets will be bound to VWF and be cleared from circulation,47 and interestingly, the bleeding score correlates with platelet count in PT-VWD.46 To this regard, it is worth noting that the patient we report here had a persistently normal platelet count, different from typical PT-VWD, who have thrombocytopenia, sometimes fluctuating.
The observation that in our study a GPIbα variant initially classified as VUS was later classified as pathogenic shows that in the presence of an uncommon bleeding phenotype and of a novel variant in a candidate gene, even with borderline laboratory test results, it is worth carrying out functional tests to explore its possible pathogenicity. The patient finally received a diagnosis of PT-VWD with mild phenotype, and correct diagnosis of PT-VWD is critical for treatment decisions because administration of VWF/FVIII concentrates or desmopressin (DDAVP) may exacerbate thrombocytopenia and bleeding in PT-VWD patients.2 Although thus far he never suffered severe spontaneous bleeding or underwent surgical procedures or tooth extractions, he will be strictly monitored for bleeding events and treated appropriately in case of invasive procedures.23
In conclusion, our data show for the first time that GOF variants outside the GPIbα C-terminal disulfide loop may be pathogenic, leading to a PT-VWD clinical phenotype. Moreover, aminoacidic changes in the LRR may cause conformational changes in the C-terminal disulfide loop of GPIbα, inducing a conformation with high affinity for VWF. These findings unravel for the first time a complex mechanism regulating the affinity of the ligand-binding region of GPIbα for VWF, a key feature in many pathological and physiological contexts, including type 2B-VWD and thrombotic thrombocytopenic purpura, and provide a new framework for future investigations of functions and regulations of the GPIb/IX/V complex.
Acknowledgments
This work was supported by a Telethon grant (GGP15063) to P.G. and by a fellowship from Fondazione Umberto Veronesi to L.B. and E.F. and by the National Institutes of Health, Heart, Lung and Blood Institute (HL146508) to M.A. The authors thank A. Savoia (IRCCS “Burlo Garofolo,” Trieste, Italy) and Francois Lanza (UMR S_949 - EFS-Alsace, Strasbourg, France) for providing the CHO β/IX cells, Luigi De Marco (Department of Translational Research, National Cancer Center, Aviano, Italy) for providing the LJ-P19 antibody and human purified VWF, and Jose Rivera (University of Murcia, Murcia, Spain) for performing high-throughput-sequencing on patient DNA.
Authorship
Contribution: L.B., E.F., A.M.M., G.G., H.K.B., M.A., L.M.-T., and A.T. performed experiments and analyzed and interpreted data; P.G. contributed the patient for the study and designed and supervised the study; L.B., M.A., A.T., and P.G. wrote the manuscript; and P.G., A.T., and M.A. critically revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Paolo Gresele, Department of Medicine and Surgery, Division of Internal and Cardiovascular Medicine, University of Perugia, Centro Didattico Edificio B piano 1, 06132 Perugia, Italy; e-mail: paolo.gresele@unipg.it.

