

Key Points
A novel type 2N VWD mouse model was established in which VWF is incapable of binding FVIII but is otherwise fully functional.
VWF2N/2N mice exhibited a severe bleeding phenotype after tail tip amputation but not in lateral tail vein or ventral artery injury models.

Abstract
Type 2N von Willebrand disease is caused by mutations in the factor VIII (FVIII) binding site of von Willebrand factor (VWF), resulting in dysfunctional VWF with defective binding capacity for FVIII. We developed a novel type 2N mouse model using CRISPR/Cas9 technology. In homozygous VWF2N/2N mice, plasma VWF levels were normal (1167 ± 257 mU/mL), but the VWF was completely incapable of binding FVIII, resulting in 53 ± 23 mU/mL of plasma FVIII levels that were similar to those in VWF-deficient (VWF−/−) mice. When wild-type human or mouse VWF was infused into VWF2N/2N mice, endogenous plasma FVIII was restored, peaking at 4 to 6 hours post-infusion, demonstrating that FVIII expressed in VWF2N mice is viable but short-lived unprotected in plasma due to dysfunctional 2N VWF. The whole blood clotting time and thrombin generation were impaired in VWF2N/2N but not in VWF−/− mice. Bleeding time and blood loss in VWF2N/2N mice were similar to wild-type mice in the lateral tail vein or ventral artery injury model. However, VWF2N/2N mice, but not VWF−/− mice, lost a significant amount of blood during the primary bleeding phase after a tail tip amputation injury model, indicating that alternative pathways can at least partially restore hemostasis when VWF is absent. In summary, we have developed a novel mouse model by gene editing with both the pathophysiology and clinical phenotype found in severe type 2N patients. This unique model can be used to investigate the biological properties of VWF/FVIII association in hemostasis and beyond.
Introduction
In blood circulation, factor VIII (FVIII) binds von Willebrand factor (VWF) noncovalently, forming a VWF/FVIII complex that contains a 50:1 molar ratio of VWF to FVIII.1-4 VWF is assembled into large multimeric structures intracellularly before being secreted into blood circulation.5-8 This bulk mass alone may be sufficient to protect FVIII from plasma protease degradation, given that the intact VWF/FVIII association is critical in maintaining the functional bioavailability of FVIII in blood circulation. Besides associating with FVIII in circulation, VWF interacts with platelets and collagen through different domains when the vessel wall is injured.9 VWF plays a fundamental role in primary hemostasis by tethering platelets at sites of injury through binding to platelet GPIbα and in secondary hemostasis by reinforcing the clot formation via binding to GPIIb/IIIa on activated platelets and collagen in the subendothelial matrix.10-14
von Willebrand disease (VWD) is caused by either an inherited deficiency of VWF protein or synthesis of a dysfunctional VWF.15 One of the important functions of VWF is to serve as a carrier protein for FVIII, protecting FVIII from proteinase degradation.4,16-18 A variant form of VWF was first identified by Montgomery and colleagues19 in which VWF binding capacity for FVIII was defective, resulting in FVIII’s rapid clearance and a phenotype similar to moderate hemophilia and subsequently referred to as type 2N VWD (2N VWD).20,21 2N VWD is caused by autosomal recessive variants of VWF in which mutations in the D′D3 region of VWF cause decreased or absent binding of FVIII to VWF.20,22-26 These individuals may have a 2N mutation on each of their VWF alleles or one 2N allele and a second allele containing a null mutation (producing no wild-type VWF).27 In each of these scenarios, only functionally abnormal VWF is synthesized, resulting in reduced or absent FVIII binding, rapid FVIII clearance, and a marked reduction in plasma FVIII. Although Swystun and coworkers28 have demonstrated 2N VWF expression by hydrodynamic tail vein injection in a VWF−/− mouse, their approach represents an ectopic hepatocyte-expression model of plasma 2N VWF dysfunction but does not recapitulate human 2N VWD in which the abnormal VWF is present not only in plasma but also in endothelial cells (ECs) and platelets. Thus, it is desirable to develop a 2N model in mice that simulates human 2N VWD characteristics in plasma and in situ in platelets and ECs. Such a model will allow us to develop a better understanding of the pathophysiology of VWD and the biological interaction between VWF and FVIII.
In this study, we used a CRISPR/Cas9 strategy to generate the first VWF2N mouse model by introducing a 2354G>A [G785E] mutation that is known to cause human 2N VWD.26,29 We investigated how this VWF2N variant impacts FVIII function in various genetic models. Functional hemostatic properties in model mice were analyzed by rotational thromboelastometry (ROTEM) and native whole blood thrombin generation assay (nWB-TGA). Three in vivo injury models were used to assess how VWF2N affects hemostasis in vein, artery, or severed tail tip transection (TTT) injury.
Materials and methods
Antibodies and reagents
The details of antibodies and reagents as well as the methods and statistics used in this study are provided in supplemental Data.
Mice
All mice used in this study were in the C57BL/6J background. All animal studies were performed according to protocols approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin. Isoflurane or ketamine was used for anesthesia.
Generation of type 2N VWD mouse models
2354G>a [G785E], a known human 2N VWF variant,26 was introduced into cloned mouse VWF (mVWF) complementary DNA (cDNA), and expression was confirmed in HEK293T cells. A CRISPR-Cas9 strategy was used to generate a 2N VWD mouse model. A guide RNA (gRNA) targeting exon 18 of the mouse Vwf gene was cloned into a plasmid that expresses both the gRNA and Cas9. As shown in Figure 1A, an asymmetric single-stranded oligodeoxynucleotide homology-directed repair template spanning the predicted Cas9 cleavage site was designed to introduce 3 single nucleotide changes into the Vwf gene following procedures as reported.30 The gRNA/Cas9 plasmid and homology-directed repair template were injected into C57BL/6J mouse zygotes. The resulting founders were bred with C57BL/6J mice to establish 2N VWD lineages. Heterozygous VWF2N/+ offspring were crossed to generate the homozygous VWF2N/2N model as well as additional VWF2N/+ and wild-type VWF+/+ controls. VWF2N/2N mice were crossed with VWF−/− mice31 to generate VWF2N/− mice.
![Establishing the type 2N VWD mouse model using a CRISPR/Cas9 strategy. (A) A gRNA and asymmetric single-stranded oligodeoxynucleotide homology-directed repair template spanning the predicted Cas9 cleavage site were designed to introduce 3 single nucleotide changes into the mouse Vwf gene. The 2354G>a [G785E] mutation causes type 2N VWD. The other 2 changes are silent mutations, 2346G>a and 2358G>c, which eliminate the Cas9-requisite protospacer adjacent motif sequence and create a diagnostic Xho I site for genotyping, respectively. (B) Schematic diagram of genotyping by PCR and Xho I restriction enzyme digestion. (C) PCR detection of the 2354G>a variant mouse Vwf gene for genotyping. DNA was purified from peripheral blood leukocytes, and a 796-bp fragment was amplified from the mouse Vwf gene. PCR product was digested with Xho I and run on 1.25% gel. A single uncut fragment of 796 bp is observed for the wild-type mouse Vwf PCR product because it does not contain an Xho I restriction enzyme site. Two fragments (585 and 211 bp) result from the mutated type 2N mouse Vwf gene after Xho I digestion. Thus, a VWF+/+ genotype results in a single 796-bp band, VWF2N/2N genotype is indicated by 585- and 211-bp bands, and VWF2N/+ mice exhibited 796-, 585-, and 211-bp bands.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/9/10.1182_bloodadvances.2021006353/5/m_advancesadv2021006353f1.png?Expires=1769327270&Signature=AqgvuC6zqV5BB8aKSGmuOBCO-0Y~hU5-sSbCCKey9vExqf1piRitkG6B70C72fGIPftnpY1N9m3VA85~BwCbObZK2gwijtGt4~EEGM-6iTCtgmpPr61viCaCFQyaaaY7Hw2y9yGQvl71QPwSkYrAR2A-Xigslfa2D8sYOs0NA9r0to78plGjWO1iPtS3SCSfpLPWEwno7NItTUkN1zw8yblE~2IAbhRLcG5sLxRgjMJvmVPZhdS6UrGDJrS1ZxF48c8lBRl-ex2InI5vSVG1bLqVZaVLJm16zK87sJQcAwyPhtN6-lbzJiyUYvtMQdNqf7UjTXMnXLNH23T9RdvSKg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Establishing the type 2N VWD mouse model using a CRISPR/Cas9 strategy. (A) A gRNA and asymmetric single-stranded oligodeoxynucleotide homology-directed repair template spanning the predicted Cas9 cleavage site were designed to introduce 3 single nucleotide changes into the mouse Vwf gene. The 2354G>a [G785E] mutation causes type 2N VWD. The other 2 changes are silent mutations, 2346G>a and 2358G>c, which eliminate the Cas9-requisite protospacer adjacent motif sequence and create a diagnostic Xho I site for genotyping, respectively. (B) Schematic diagram of genotyping by PCR and Xho I restriction enzyme digestion. (C) PCR detection of the 2354G>a variant mouse Vwf gene for genotyping. DNA was purified from peripheral blood leukocytes, and a 796-bp fragment was amplified from the mouse Vwf gene. PCR product was digested with Xho I and run on 1.25% gel. A single uncut fragment of 796 bp is observed for the wild-type mouse Vwf PCR product because it does not contain an Xho I restriction enzyme site. Two fragments (585 and 211 bp) result from the mutated type 2N mouse Vwf gene after Xho I digestion. Thus, a VWF+/+ genotype results in a single 796-bp band, VWF2N/2N genotype is indicated by 585- and 211-bp bands, and VWF2N/+ mice exhibited 796-, 585-, and 211-bp bands.
Establishing the type 2N VWD mouse model using a CRISPR/Cas9 strategy. (A) A gRNA and asymmetric single-stranded oligodeoxynucleotide homology-directed repair template spanning the predicted Cas9 cleavage site were designed to introduce 3 single nucleotide changes into the mouse Vwf gene. The 2354G>a [G785E] mutation causes type 2N VWD. The other 2 changes are silent mutations, 2346G>a and 2358G>c, which eliminate the Cas9-requisite protospacer adjacent motif sequence and create a diagnostic Xho I site for genotyping, respectively. (B) Schematic diagram of genotyping by PCR and Xho I restriction enzyme digestion. (C) PCR detection of the 2354G>a variant mouse Vwf gene for genotyping. DNA was purified from peripheral blood leukocytes, and a 796-bp fragment was amplified from the mouse Vwf gene. PCR product was digested with Xho I and run on 1.25% gel. A single uncut fragment of 796 bp is observed for the wild-type mouse Vwf PCR product because it does not contain an Xho I restriction enzyme site. Two fragments (585 and 211 bp) result from the mutated type 2N mouse Vwf gene after Xho I digestion. Thus, a VWF+/+ genotype results in a single 796-bp band, VWF2N/2N genotype is indicated by 585- and 211-bp bands, and VWF2N/+ mice exhibited 796-, 585-, and 211-bp bands.
VWF and FVIII assays
Blood samples were collected by tail bleed, and plasma was isolated as reported.18 Functional FVIII activity (FVIII:C) levels were quantified by a modified chromogenic assay as previously described.32,33
VWF antigen (VWF:Ag) levels in mouse plasma were determined by enzyme-linked immunosorbent assay (ELISA) as described in our previous report.34 Pooled plasma from C57BL/6J mice was used as the standard. For human VWF:Ag ELISA, anti-human VWF monoclonal antibodies were used. Pooled plasma from healthy human individuals was used as the standard. VWF binding to FVIII was determined by measuring VWF binding to antibody-captured FVIII or, conversely, FVIII binding to antibody-captured VWF.
To determine the biological functions of 2N VWF in binding to collagen and platelets as well as in multimerization, we performed bindings assays for VWF/collagen-III, VWF/collagen-IV, VWF/platelets, and VWF multimers using samples collected from VWF2N/2N mice.
To investigate whether VWF with normal FVIII binding capacity can restore endogenous plasma FVIII in VWF2N mice, recombinant human VWF (rhVWF) or mVWF (in FVIII−/− mouse plasmas) were infused into VWF2N/2N mice. Blood samples were collected at various time points after infusion, and plasmas were isolated for VWF and FVIII assays.
Phenotypic assessments
The bleeding phenotype in VWF2N mice was assessed by in vitro ROTEM assay and nWB-TGA following procedures described in our previous reports35,36 and by 3 in vivo injury models: (1) lateral tail vein transection (TVT) injury,28,37 ,38 (2) ventral tail artery transection (TAT) injury, and (3) TTT injury. All in vivo bleeding assays were blinded. Primary and rechallenge bleeding times were recorded, and blood loss was quantified.
Statistical analysis
Data are presented as the mean ± standard deviation. Statistical analysis was performed using GraphPad Prism 7 (GraphPad Software, La Jolla, CA) and SigmaPlot 14.0 (Systat Software, Inc., San Jose, CA). A value of P < .05 was considered statistically significant.
Results
Establishing VWF2N models
In preliminary studies, 8 different human 2N mutations were introduced into cloned mVWF cDNA and expressed in HEK293T cells. The binding of each of the 2N mutants to both murine and human FVIII was tested. The G785E 2N variant had the most severe defect with negligible binding to both rhFVIII and recombinant mouse FVIII (supplemental Figure 1A-B) and was therefore chosen for mouse model study. Using CRISPR/Cas9 strategy, we generated 2 lines of VWF2N mice (referred to as VWF2N1 and VWF2N2) with the human 2N VWD-causative 2354G>A [G785E] mutation in a C57BL/6J background. Animals were screened by polymerase chain reaction (PCR) genotyping. As shown in Figure 1A-B, a diagnostic Xho I site was introduced into the 2N VWF allele. Genotyping was performed by PCR amplification of the exon 18 region, followed by Xho I digestion. A 796-bp band was detected from the wild-type VWF allele. Two bands, 585 and 211 bp, were detected from the allele containing the VWF2N mutation. Thus, for VWF+/+ mice, only the 796-bp fragment was detected, and for homozygous VWF2N (VWF2N/2N) mice, 585- and 211-bp fragments were detected. Heterozygous animals (VWF2N/+) displayed 3 fragments (Figure 1C).
To characterize VWF2N animals, we used an ELISA18,34,39 to determine plasma VWF:Ag levels and a chromogenic assay18,32,34 to measure FVIII:C levels. The plasma levels of VWF:Ag in VWF2N/2N mice of both lines (1167 ± 257 [n = 31] and 1169 ± 319 mU/mL [n = 20]) were similar to those in VWF2N/+ mice (1023 ± 296 [n = 44] and 1029 ± 348 mU/mL [n = 29]) and VWF+/+ littermates [1089 ± 309 and 1040 ± 212 mU/mL, respectively) (Figure 2A). We also performed VWF propeptide (VWFpp) (data not shown) and used the ratio of VWFpp/VWF:Ag to determine the quality of sample collected. All data included in our report were from samples with VWFpp/VWF:Ag ≤ 3.0 because the ratio of VWFpp/VWF:Ag > 3 is an indicator of sample activation. Of note, the plasma FVIII level in VWF2N/2N mice was severely decreased (53 ± 23 mU/mL in VWF2N1/2N1 and 35 ± 29 mU/mL in VWF2N2/2N2), whereas VWF2N/+ had FVIII levels similar to VWF+/+ mice (Figure 2B). When we performed VWF/FVIII binding assays, no plasma VWF from VWF2N/2N mice was bound to anti-FVIII antibody-captured recombinant human full-length (rhf) FVIII. In contrast, 99.5% ± 8.1% of VWF from VWF+/+ mice and 56.2% ± 7.9% from VWF2N/+ mice could bind to rhfFVIII (Figure 2C-D). Likewise, no endogenous mouse FVIII was bound to plasma VWF in VWF2N/2N mice from the VWF2N1 or VWF2N2 colony in VWF/FVIII co-capture assays (supplemental Figure 2). Both VWF2N lineages displayed similar characteristics of VWF and FVIII expression in plasma. Furthermore, the functions of VWF from VWF2N/2N mice in binding to collagen and platelets and in multimerization were comparable to those in VWF+/+ littermates (Figure 3A-D). Together, these data confirm that our VWF2N mouse model has normal levels of VWF in plasma but undetectable binding of FVIII to VWF and otherwise is fully functional. Thus, we maintained the VWF2N1 line, henceforth referred to as simply VWF2N, and animals derived from this line were used for the remaining studies.
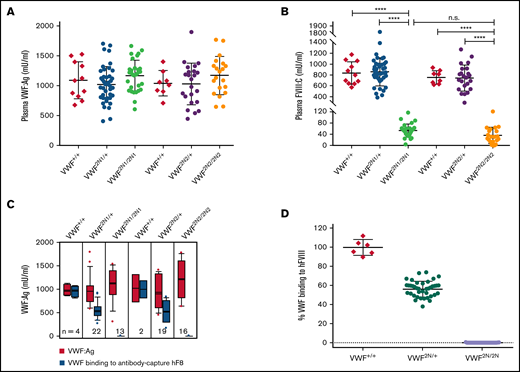
Characterization of VWF and FVIII expression in type 2N VWD model mice. Blood samples were collected from 2 lines of VWF2N model mice by tail bleeds using 3.8% sodium citrate as an anticoagulant, and plasmas were isolated for VWF and FVIII assays. Plasmas from VWF+/+ littermates were used as controls in parallel. (A) Plasma VWF antigen (VWF:Ag) levels. Mouse VWF antigen (VWF:Ag) levels were determined by ELISA using anti-mVWF monoclonal antibody 344.2 for capture and biotin-conjugated monoclonal antibody 332.2 for detection. Plasma pooled from our wild-type C57BL/6J colony was used as the standard. (B) Plasma functional FVIII activity (FVIII:C) levels. Plasma FVIII:C levels were determined by a chromogenic assay. Recombinant human B-domain deleted FVIII (rhFVIII, Xyntha) was used as the standard. (C) The capacity of VWF binding to human FVIII. Anti–human FVIII monoclonal antibody 103.1 was coated on a 96-well plate, and rhfFVIII (Kogenate) was captured from a 1-U/mL solution. Plasmas from VWF2N mice were incubated with the antibody captured rhfF8, unbound mVWF was washed off, and the remaining FVIII-bound mVWF was detected using mVWF ELISA detection reagents. The standard curve was constructed by measuring mVWF binding from serially diluted pooled plasma from our wild-type C57BL/6J colony. (D) The percentage of mVWF capable of binding to captured hFVIII. Data from 2N1 and 2N2 colonies were combined for this analysis. The percentage was calculated by dividing the level of VWF binding to antibody-captured hFVIII by the plasma VWF level in the same animal (using data from Figure 2C). ****P < .0001. n.s., no statistically significant difference between 2 groups. These results demonstrate that VWF2N mice have normal levels of plasma VWF but are incapable of binding FVIII, resulting in severely reduced levels of plasma FVIII:C.
Characterization of VWF and FVIII expression in type 2N VWD model mice. Blood samples were collected from 2 lines of VWF2N model mice by tail bleeds using 3.8% sodium citrate as an anticoagulant, and plasmas were isolated for VWF and FVIII assays. Plasmas from VWF+/+ littermates were used as controls in parallel. (A) Plasma VWF antigen (VWF:Ag) levels. Mouse VWF antigen (VWF:Ag) levels were determined by ELISA using anti-mVWF monoclonal antibody 344.2 for capture and biotin-conjugated monoclonal antibody 332.2 for detection. Plasma pooled from our wild-type C57BL/6J colony was used as the standard. (B) Plasma functional FVIII activity (FVIII:C) levels. Plasma FVIII:C levels were determined by a chromogenic assay. Recombinant human B-domain deleted FVIII (rhFVIII, Xyntha) was used as the standard. (C) The capacity of VWF binding to human FVIII. Anti–human FVIII monoclonal antibody 103.1 was coated on a 96-well plate, and rhfFVIII (Kogenate) was captured from a 1-U/mL solution. Plasmas from VWF2N mice were incubated with the antibody captured rhfF8, unbound mVWF was washed off, and the remaining FVIII-bound mVWF was detected using mVWF ELISA detection reagents. The standard curve was constructed by measuring mVWF binding from serially diluted pooled plasma from our wild-type C57BL/6J colony. (D) The percentage of mVWF capable of binding to captured hFVIII. Data from 2N1 and 2N2 colonies were combined for this analysis. The percentage was calculated by dividing the level of VWF binding to antibody-captured hFVIII by the plasma VWF level in the same animal (using data from Figure 2C). ****P < .0001. n.s., no statistically significant difference between 2 groups. These results demonstrate that VWF2N mice have normal levels of plasma VWF but are incapable of binding FVIII, resulting in severely reduced levels of plasma FVIII:C.
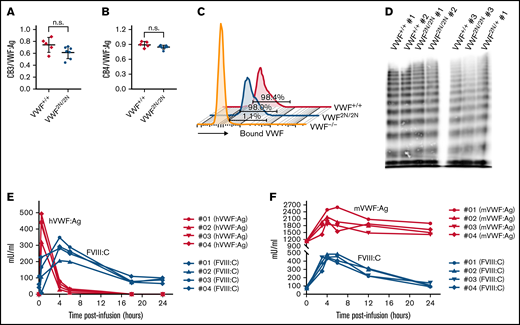
Evaluation of the functional properties of 2N VWF and the viability of plasma FVIII activity in VWF2N/2N mice. (A-B) The capacity of 2N VWF in collagen binding. To investigate if VWF in 2N mice can function normally in binding to collagen, we performed collagen III and IV binding assays, in which a 96-well plate was coated with collagen and plasma VWF:Ag bound to collagen was determined using a protocol similar to VWF:Ag ELISA. Plasma VWF:Ag levels were determined by ELISA in parallel. Pooled plasma from wild-type C57BL/6J mice was used as a standard. (A) The ratio of collagen III-bound VWF (CB3) to plasma VWF:Ag. (B) The ratio of collagen IV-bound VWF (CB4) to plasma VWF:Ag. (C) The capacity of 2N VWF in platelet binding. To determine if VWF in 2N mice can bind effectively to platelets, we performed a VWF/platelet-binding assay. VWF was activated by botrocetin in whole blood, and VWF-bound platelets were analyzed by flow cytometry. Samples from VWF−/− and VWF+/+ mice were used as controls in parallel. Data shown are representative histograms and the mean value from 3 mice in each group. (D) The ability of 2N VWF in multimerization. To examine if VWF in 2N mice can fully multimerize, we ran VWF multimers on plasma samples from 2N mice. Plasmas from VWF2N/+ and VWF+/+ mice were run in parallel. (E-F) The viability of plasma FVIII in 2N mice. To investigate if FVIII is viable in VWF2N/2N mice, rhVWF, or mVWF, was infused into VWF2N/2N mice. Blood samples were collected at various time points after infusion, and plasmas were isolated for VWF and FVIII assays. (E) Functional FVIII:C levels in VWF2N/2N mice upon rhVWF infusion. VWF2N/2N mice were infused with 50 U/kg of rhVWF (Vonvendi, Baxalta) via retro-orbital venous plexus injection. Human VWF:Ag levels were determined by VWF:Ag ELISA using anti-human specific antibodies, and pooled human plasma was used as the standard. Plasma FVIII:C levels were determined by a chromogenic assay, and rhF8 was used as the standard. (F) Functional FVIII:C levels in VWF2N/2N mice upon mVWF infusion. VWF2N/2N mice were infused with 200 μL of pooled plasma from FVIII−/− mice. Mouse VWF:Ag levels were determined by ELISA using mouse-specific antibodies, and pooled plasma from wild-type C57BL6 mice was used as the standard. Plasma FVIII:C levels were determined by a chromogenic assay, and rhF8 was used as the standard. These data demonstrate that VWF from 2N mice has normal functional activities in binding to collagen and platelets and in multimerization. The endogenous mouse FVIII in VWF2N/2N mice is bioavailable and can be stabilized in plasma in the presence of normal VWF.
Evaluation of the functional properties of 2N VWF and the viability of plasma FVIII activity in VWF2N/2N mice. (A-B) The capacity of 2N VWF in collagen binding. To investigate if VWF in 2N mice can function normally in binding to collagen, we performed collagen III and IV binding assays, in which a 96-well plate was coated with collagen and plasma VWF:Ag bound to collagen was determined using a protocol similar to VWF:Ag ELISA. Plasma VWF:Ag levels were determined by ELISA in parallel. Pooled plasma from wild-type C57BL/6J mice was used as a standard. (A) The ratio of collagen III-bound VWF (CB3) to plasma VWF:Ag. (B) The ratio of collagen IV-bound VWF (CB4) to plasma VWF:Ag. (C) The capacity of 2N VWF in platelet binding. To determine if VWF in 2N mice can bind effectively to platelets, we performed a VWF/platelet-binding assay. VWF was activated by botrocetin in whole blood, and VWF-bound platelets were analyzed by flow cytometry. Samples from VWF−/− and VWF+/+ mice were used as controls in parallel. Data shown are representative histograms and the mean value from 3 mice in each group. (D) The ability of 2N VWF in multimerization. To examine if VWF in 2N mice can fully multimerize, we ran VWF multimers on plasma samples from 2N mice. Plasmas from VWF2N/+ and VWF+/+ mice were run in parallel. (E-F) The viability of plasma FVIII in 2N mice. To investigate if FVIII is viable in VWF2N/2N mice, rhVWF, or mVWF, was infused into VWF2N/2N mice. Blood samples were collected at various time points after infusion, and plasmas were isolated for VWF and FVIII assays. (E) Functional FVIII:C levels in VWF2N/2N mice upon rhVWF infusion. VWF2N/2N mice were infused with 50 U/kg of rhVWF (Vonvendi, Baxalta) via retro-orbital venous plexus injection. Human VWF:Ag levels were determined by VWF:Ag ELISA using anti-human specific antibodies, and pooled human plasma was used as the standard. Plasma FVIII:C levels were determined by a chromogenic assay, and rhF8 was used as the standard. (F) Functional FVIII:C levels in VWF2N/2N mice upon mVWF infusion. VWF2N/2N mice were infused with 200 μL of pooled plasma from FVIII−/− mice. Mouse VWF:Ag levels were determined by ELISA using mouse-specific antibodies, and pooled plasma from wild-type C57BL6 mice was used as the standard. Plasma FVIII:C levels were determined by a chromogenic assay, and rhF8 was used as the standard. These data demonstrate that VWF from 2N mice has normal functional activities in binding to collagen and platelets and in multimerization. The endogenous mouse FVIII in VWF2N/2N mice is bioavailable and can be stabilized in plasma in the presence of normal VWF.
The effect of VWF on FVIII expression in various VWF deficient models
To examine whether FVIII expressed in VWF2N mice is viable but short-lived due to lack of protection by 2N VWF in plasma, normal VWF was infused into VWF2N/2N mice, and plasmas were collected for FVIII:C and VWF:Ag assays. As shown in Figure 3E, infusion of 50 U/kg rhVWF into VWF2N/2N mice restored endogenous mouse FVIII:C in plasma, peaking at 4 hours postinfusion with a level of 282 ± 58 mU/mL. When 200 μL of pooled plasma from FVIII−/− mice, which contains normal mVWF, was infused into VWF2N/2N mice, plasma FVIII:C was restored to 451 ± 23 mU/mL at 4 hours (Figure 3F). These results demonstrate that endogenous mouse FVIII in VWF2N/2N mice is bioavailable and can be restored and stabilized in plasma in the presence of normal VWF.
To mimic the VWD type 2N/null compound heterozygous phenotype40,41 in mice, we crossed VWF2N/2N with VWF−/− mice to get VWF2N/− mice and compared plasma VWF:Ag and FVIII:C levels in mouse models of various genotypes. As shown in Figure 4A, plasma VWF:Ag levels in VWF2N/− mice were 544 ± 200 mU/mL, which were significantly lower than those in VWF2N/+, VWF2N/2N, and VWF+/+ mice. Plasma FVIII:C levels in VWF2N/− mice were 58 ± 33 mU/mL, comparable to VWF2N/2N (56 ± 19 mU/mL) and VWF−/− (79 ± 34 mU/mL) mice, but markedly reduced compared with VWF2N/+, VWF+/−, and VWF+/+ mice (Figure 4B). The FVIII:C level in the VWF2N/+ group was not significantly different from the VWF+/+ group. The FVIII:C level in the VWF+/− group was significantly lower compared with the VWF+/+ and VWF2N/+ groups but higher than the VWF−/− group (Figure 4B). These data demonstrate that the viability of FVIII in plasma in VWD model mice is governed by its association or inability to associate with VWF.
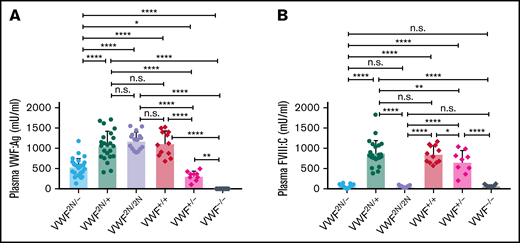
The impact of VWF on functional plasma FVIII:C levels in various mouse models of VWD. We compared how VWF impacts plasma FVIII:C expression levels in VWF2N/−, VWF2N/+, VWF2N/2N, VWF+/+ (littermates), VWF+/−, and VWF−/− mice. We crossed VWF2N/2N with VWF−/− mice to generate compound heterozygous VWF2N/- mice. Blood samples were collected from mice via retro-orbital venous plexus bleeds using 3.8% sodium citrate as an anticoagulant. Plasma VWF:Ag levels were determined by ELISA using anti-mVWF specific antibodies, and pooled plasma from wild-type C57BL/6J mice was used as the standard. Plasma FVIII:C levels were determined by a chromogenic assay, and rhF8 was used as the standard. (A) Plasma VWF:Ag levels. (B) Plasma FVIII:C levels. *P < .05; **P < .01; ***P < .001; ****P < .0001. n.s., no statistically significant difference between the 2 groups. These data demonstrate that the viability of plasma FVIII:C in VWD model mice is governed by its association with or inability to associate with VWF.
The impact of VWF on functional plasma FVIII:C levels in various mouse models of VWD. We compared how VWF impacts plasma FVIII:C expression levels in VWF2N/−, VWF2N/+, VWF2N/2N, VWF+/+ (littermates), VWF+/−, and VWF−/− mice. We crossed VWF2N/2N with VWF−/− mice to generate compound heterozygous VWF2N/- mice. Blood samples were collected from mice via retro-orbital venous plexus bleeds using 3.8% sodium citrate as an anticoagulant. Plasma VWF:Ag levels were determined by ELISA using anti-mVWF specific antibodies, and pooled plasma from wild-type C57BL/6J mice was used as the standard. Plasma FVIII:C levels were determined by a chromogenic assay, and rhF8 was used as the standard. (A) Plasma VWF:Ag levels. (B) Plasma FVIII:C levels. *P < .05; **P < .01; ***P < .001; ****P < .0001. n.s., no statistically significant difference between the 2 groups. These data demonstrate that the viability of plasma FVIII:C in VWD model mice is governed by its association with or inability to associate with VWF.
Assessment of the functional hemostatic properties in whole blood of VWF2N model mice
To determine how VWF2N impacts hemostatic properties in whole blood, we used ROTEM and nWB-TGA. As shown in Figure 5A, ROTEM analysis showed that the whole blood clotting time (CT) in VWF2N/2N and VWF2N/− groups (10.5 ± 1.13 and 11.42 ± 1.01 minutes, respectively) were significantly longer than in the C57BL/6J group (8.68 ± 0.72 minutes). The clot formation time in the VWF2N/− group was significantly longer, and α-angle was smaller compared with the C57BL/6J group. There were no statistically significant differences between other groups in other parameters, and there was no statistically significant difference between the VWF−/− and C57BL/6J groups in all parameters (Figure 5A).
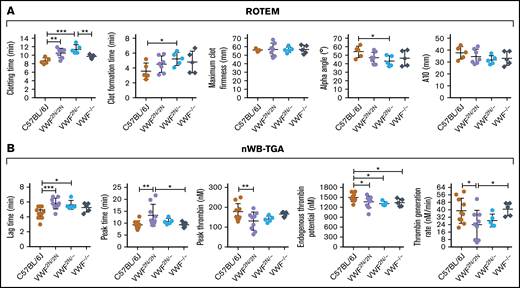
Assessment of the biological hemostatic properties in VWF2N mice whole blood. Blood samples were collected from the vena cava (terminal experiment) using 3.8% sodium citrate as an anticoagulant (vol/vol 1:10) and analyzed by nonactivated ROTEM analysis and nWB-TGA. (A) ROTEM analysis of whole blood. ROTEM standard cups were preloaded with 21 µL of 0.2M CaCl2, and then 300 µL of whole blood was added. Clot formation was recorded using the NATEM measurement until maximum clot firmness reached its peak. (B) nWB-TGA analysis of whole blood. Fifteen microliters of whole blood was recalcified in the presence of a rhodamine-based, thrombin-cleavable, fluorescent substrate and added in duplicate to filter paper placed within the wells of a black 96-well plate. Change in fluorescence was measured over time and converted to thrombin generation. Conversions were calculated from a curve generated during a calibration experiment using a thrombin standard. C57BL/6J and VWF−/− mice served as controls. *P < .05; **P < .01; ***P < .001. These data demonstrate that functional hemostatic properties in VWF2N/2N whole blood are defective.
Assessment of the biological hemostatic properties in VWF2N mice whole blood. Blood samples were collected from the vena cava (terminal experiment) using 3.8% sodium citrate as an anticoagulant (vol/vol 1:10) and analyzed by nonactivated ROTEM analysis and nWB-TGA. (A) ROTEM analysis of whole blood. ROTEM standard cups were preloaded with 21 µL of 0.2M CaCl2, and then 300 µL of whole blood was added. Clot formation was recorded using the NATEM measurement until maximum clot firmness reached its peak. (B) nWB-TGA analysis of whole blood. Fifteen microliters of whole blood was recalcified in the presence of a rhodamine-based, thrombin-cleavable, fluorescent substrate and added in duplicate to filter paper placed within the wells of a black 96-well plate. Change in fluorescence was measured over time and converted to thrombin generation. Conversions were calculated from a curve generated during a calibration experiment using a thrombin standard. C57BL/6J and VWF−/− mice served as controls. *P < .05; **P < .01; ***P < .001. These data demonstrate that functional hemostatic properties in VWF2N/2N whole blood are defective.
When hemostatic properties were assessed by nWB-TGA, the lag time and peak time in the VWF2N/2N group were 5.78 ± 0.72 and 13.36 ± 4.54 minutes, respectively, which were significantly longer than the C57BL/6J group (4.54 ± 0.79 and 9.38 ± 1.66 minutes, respectively). The peak thrombin, endogenous thrombin potential (ETP), and thrombin generation rate in the VWF2N/2N group were significantly lower than those in the C57BL/6J group (Figure 5B). The lag time was longer, and ETP was lower in the VWF2N/− group compared with the C57BL6 group, but other parameters were comparable between the 2 groups. In the VWF−/− group, ETP was the only parameter that was significantly different when compared with the C57BL/6J group. Interestingly, the peak time was longer, and thrombin generation rate was lower in the VWF2N/2N group compared with the VWF−/− group (Figure 5B).
Together, these data demonstrate that functional hemostatic properties in VWF2N/2N whole blood were defective as determined by ROTEM and nWB-TGA.
Assessment of the in vivo bleeding phenotype in VWF2N model mice
To examine how 2N VWF impacts the bleeding phenotype in mice, we used blinded in vivo TVT, TAT, and TTT injury experiments. In the TVT model, there were no significant differences in primary bleeding time, or blood loss among groups of VWF2N/2N, VWF2N/+, VWF2N/−, VWF+/−, VWF−/−, VWF+/+ (littermates of VWF2N), and C57BL/6J mice. Primary bleeding times and blood loss ranged from 1.1 ± 0.2 to 2.5 ± 1.1 minutes and 12.9 ± 6.9 to 37.9 ± 31.1 μL, respectively. The rechallenge bleeding time and blood lost in VWF2N model mice, including the VWF2N/2N, VWF2N/+, and VWF2N/− groups, were comparable to those obtained in VWF+/+, VWF+/−, and C57BL/6J mice. However, the rechallenge bleeding time in the VWF−/− group (23.4 ± 16.5 minutes) was significantly longer than all other groups (2.6 ± 0.3 to 3.0 ± 1.2 minutes). Likewise, the rechallenge blood loss in the VWF−/− group (183.7 ± 196.9 μL) was significantly higher than in the other groups (11.2 ± 5.5 to 27.4 ± 17.1 μL) (Figure 6A-B;,Table 1).
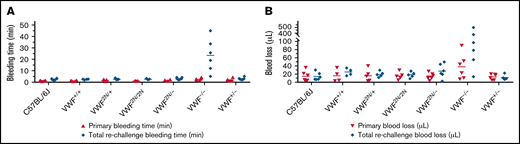
Assessment of the bleeding phenotype in VWF2N mice using lateral TVT injury model. Male and female mice with ages of 8-20 weeks old of VWF+/+, VWF2N/+, VWF2N/2N, VWF2N/-, VWF−/−, and VWF+/− genotypes were used in this study. Animals were anesthetized with isoflurane, and lateral TVT was performed at the position of 2.5 mm diameter of the prewarmed tail, introducing a 1-mm depth incision of the left lateral tail vein by sliding a scalpel blade through a transverse groove in an aluminum transection template block. The wounded tail was submerged into 14 mL of prewarmed saline and monitored for 15 minutes. The tail was removed from the saline if the bleeding stopped within 15 minutes or at 15 minutes if it did not stop, and clotting was rechallenged a total of 3 times. Primary and rechallenge bleeding times were recorded. Blood loss was quantified by lysing red cells in 10 mL of distilled H2O and measuring hemoglobin at OD575 nm, and blood loss was calculated according to a standard curve generated from known amounts of pooled blood from wild-type C57BL/6J mice. Total bleeding times and blood losses during 3 rechallenges were combined. Mice from our wild-type C57BL/6J colony and VWF+/+ (VWF2N littermates, also on a C57BL6 background) served as controls. (A) Bleeding times during primary challenge and rechallenges. (B) Blood loss during primary challenge and rechallenges. These data demonstrate that 2N VWF can still help to initiate clot formation in vein injury if the subendothelial matrix around the wound remains.
Assessment of the bleeding phenotype in VWF2N mice using lateral TVT injury model. Male and female mice with ages of 8-20 weeks old of VWF+/+, VWF2N/+, VWF2N/2N, VWF2N/-, VWF−/−, and VWF+/− genotypes were used in this study. Animals were anesthetized with isoflurane, and lateral TVT was performed at the position of 2.5 mm diameter of the prewarmed tail, introducing a 1-mm depth incision of the left lateral tail vein by sliding a scalpel blade through a transverse groove in an aluminum transection template block. The wounded tail was submerged into 14 mL of prewarmed saline and monitored for 15 minutes. The tail was removed from the saline if the bleeding stopped within 15 minutes or at 15 minutes if it did not stop, and clotting was rechallenged a total of 3 times. Primary and rechallenge bleeding times were recorded. Blood loss was quantified by lysing red cells in 10 mL of distilled H2O and measuring hemoglobin at OD575 nm, and blood loss was calculated according to a standard curve generated from known amounts of pooled blood from wild-type C57BL/6J mice. Total bleeding times and blood losses during 3 rechallenges were combined. Mice from our wild-type C57BL/6J colony and VWF+/+ (VWF2N littermates, also on a C57BL6 background) served as controls. (A) Bleeding times during primary challenge and rechallenges. (B) Blood loss during primary challenge and rechallenges. These data demonstrate that 2N VWF can still help to initiate clot formation in vein injury if the subendothelial matrix around the wound remains.
Statistical analysis of the bleeding time and blood loss in mice with various VWF genotypes using lateral TVT bleeding test
| Two-way ANOVA multiple comparisons of mice with various VWF genotypes | Bleeding time, min | Blood loss, volume | ||
|---|---|---|---|---|
| Primary (P) | Rechallenge (P) | Primary (P) | Rechallenge (P) | |
| C57BL/6J vs VWF+/+ | .9705 | .9951 | .9756 | .7723 |
| C57BL/6J vs VWF2N/+ | .8998 | .9644 | .9715 | .8835 |
| C57BL/6J vs VWF2N/2N | .9435 | .9707 | .9903 | .9120 |
| C57BL/6J vs VWF2N/− | .9178 | .8527 | .9952 | .6933 |
| C57BL/6J vs VWF−/− | .6353 | <.0001**** | .4872 | <.0001**** |
| C57BL/6J vs VWF+/− | .7570 | .8527 | .9569 | .9275 |
| VWF+/+ vs VWF2N/+ | .8814 | .9632 | .9978 | .8838 |
| VWF+/+ vs VWF2N/2N | .9773 | .9688 | .9853 | .8582 |
| VWF+/+ vs VWF2N/− | .8972 | .8632 | .9713 | .9494 |
| VWF+/+ vs VWF−/− | .6447 | <.0001**** | .5544 | <.0001**** |
| VWF+/+ vs VWF+/− | .7537 | .8632 | .9371 | .7109 |
| VWF2N/+ vs VWF2N/2N | .8506 | .9940 | .9821 | .9725 |
| VWF2N/+ vs VWF2N/− | .9780 | .8946 | .9670 | .8184 |
| VWF2N/+ vs VWF−/− | .7440 | <.0001**** | .5306 | <.0001**** |
| VWF2N/+ vs VWF+/− | .8656 | .8946 | .9305 | .8155 |
| VWF2N/2N vs VWF2N/− | .8656 | .8884 | .9857 | .7905 |
| VWF2N/2N vs VWF−/− | .6011 | <.0001**** | .5154 | <.0001**** |
| VWF2N/2N vs VWF+/− | .7145 | .8884 | .9492 | .8436 |
| VWF2N/- vs VWF−/− | .7104 | <.0001**** | .4835 | <.0001**** |
| VWF2N/- vs VWF+/− | .8365 | >.9999 | .9617 | .6276 |
| VWF−/− vs VWF+/− | .8689 | <.0001**** | .4542 | <.0001**** |
| Two-way ANOVA multiple comparisons of mice with various VWF genotypes | Bleeding time, min | Blood loss, volume | ||
|---|---|---|---|---|
| Primary (P) | Rechallenge (P) | Primary (P) | Rechallenge (P) | |
| C57BL/6J vs VWF+/+ | .9705 | .9951 | .9756 | .7723 |
| C57BL/6J vs VWF2N/+ | .8998 | .9644 | .9715 | .8835 |
| C57BL/6J vs VWF2N/2N | .9435 | .9707 | .9903 | .9120 |
| C57BL/6J vs VWF2N/− | .9178 | .8527 | .9952 | .6933 |
| C57BL/6J vs VWF−/− | .6353 | <.0001**** | .4872 | <.0001**** |
| C57BL/6J vs VWF+/− | .7570 | .8527 | .9569 | .9275 |
| VWF+/+ vs VWF2N/+ | .8814 | .9632 | .9978 | .8838 |
| VWF+/+ vs VWF2N/2N | .9773 | .9688 | .9853 | .8582 |
| VWF+/+ vs VWF2N/− | .8972 | .8632 | .9713 | .9494 |
| VWF+/+ vs VWF−/− | .6447 | <.0001**** | .5544 | <.0001**** |
| VWF+/+ vs VWF+/− | .7537 | .8632 | .9371 | .7109 |
| VWF2N/+ vs VWF2N/2N | .8506 | .9940 | .9821 | .9725 |
| VWF2N/+ vs VWF2N/− | .9780 | .8946 | .9670 | .8184 |
| VWF2N/+ vs VWF−/− | .7440 | <.0001**** | .5306 | <.0001**** |
| VWF2N/+ vs VWF+/− | .8656 | .8946 | .9305 | .8155 |
| VWF2N/2N vs VWF2N/− | .8656 | .8884 | .9857 | .7905 |
| VWF2N/2N vs VWF−/− | .6011 | <.0001**** | .5154 | <.0001**** |
| VWF2N/2N vs VWF+/− | .7145 | .8884 | .9492 | .8436 |
| VWF2N/- vs VWF−/− | .7104 | <.0001**** | .4835 | <.0001**** |
| VWF2N/- vs VWF+/− | .8365 | >.9999 | .9617 | .6276 |
| VWF−/− vs VWF+/− | .8689 | <.0001**** | .4542 | <.0001**** |
P < .0001.
We also performed TAT injury on VWF2N/2N, VWF−/−, and C57BL/6J mice. As shown in supplemental Figure 3A-B, there were no statistically significant differences in bleeding time or blood loss regardless of primary or rechallenged bleeding between the VWF2N/2N and C57BL/6J groups. VWF−/− mice lost a considerable amount of blood, with significantly longer bleeding times and blood loss in both primary and rechallenge bleeding compared with VWF2N/2N and C57BL/6J mice. Three of 5 VWF−/− mice died during the rechallenge bleeding test, and data were recorded up to the point of animal death. Together, our data demonstrate that 2N VWF can still help to initiate clot formation in both veins and arteries if the subendothelial matrix around the wound remains.
In the TTT injury model, in which 4 mm of the tail tip was completely severed, the primary and secondary bleeding times in VWF2N/2N mice were 8.2 ± 4.1 and 8.2 ± 3.9 minutes, which were significantly longer than in VWF+/+ littermates (1.4 ± 0.8 and 0.7 ± 0.5 minutes, respectively) and C57BL/6J mice. The primary and rechallenge blood losses in VWF2N/2N mice (263.2 ± 219.8 and 155.7 ± 112 μL, respectively) were markedly more than VWF+/+ littermates (13.6 ± 9.6 and 3.1 ± 1.8 μL, respectively) and C57BL/6J mice (Figure 7A-B;,Table 2). Four of 5 mice in the VWF2N/2N group bled for the entire primary and rechallenge tests, but all mice in VWF+/+ littermates experienced clotting within 3 minutes (Figure 7A). Primary bleeding time and blood loss in the VWF−/− group were not significantly different compared with the C57BL/6J group, but VWF−/− mice bled longer and lost more blood than C57BL/6J mice during rechallenge (Figure 7A-B). There were no significant differences in primary and rechallenge bleeding times between the VWF2N/2N and VWF−/− groups.
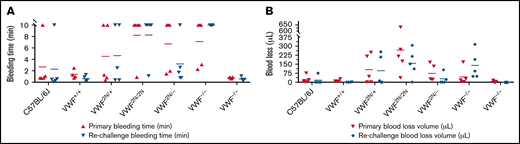
Assessment of the bleeding phenotype in VWF2N mice by a TTT injury model. Male and female mice age 8 to 20 weeks of VWF+/+, VWF2N/+, VWF2N/2N, VWF2N/−, VWF−/−, and VWF+/− genotypes were assessed for their bleeding phenotype. Animals were anesthetized with isoflurane, and 4 mm of the tail tip was clipped. The wounded tail was submerged in 14 mL of prewarmed saline and monitored for 10 minutes. The tail was removed if the bleeding stopped within 10 minutes or at 10 minutes if it did not stop and rechallenged once. Bleeding times were recorded. Blood loss was measured by lysing red cells in 10 mL of distilled H2O and measuring hemoglobin at OD575 nm, and blood loss was calculated according to a standard curve generated from known amounts of pooled blood from wild-type C57BL/6J mice. Mice from our wild-type C57BL/6J colony and VWF+/+ (VWF2N littermates, also on a C57BL6 background) served as controls. (A) Bleeding times during primary challenge and rechallenge. (B) Blood loss during primary challenge and rechallenge. These data demonstrate that both primary and secondary hemostasis are impaired in VWF2N/2N mice in a severed tail tip injury model.
Assessment of the bleeding phenotype in VWF2N mice by a TTT injury model. Male and female mice age 8 to 20 weeks of VWF+/+, VWF2N/+, VWF2N/2N, VWF2N/−, VWF−/−, and VWF+/− genotypes were assessed for their bleeding phenotype. Animals were anesthetized with isoflurane, and 4 mm of the tail tip was clipped. The wounded tail was submerged in 14 mL of prewarmed saline and monitored for 10 minutes. The tail was removed if the bleeding stopped within 10 minutes or at 10 minutes if it did not stop and rechallenged once. Bleeding times were recorded. Blood loss was measured by lysing red cells in 10 mL of distilled H2O and measuring hemoglobin at OD575 nm, and blood loss was calculated according to a standard curve generated from known amounts of pooled blood from wild-type C57BL/6J mice. Mice from our wild-type C57BL/6J colony and VWF+/+ (VWF2N littermates, also on a C57BL6 background) served as controls. (A) Bleeding times during primary challenge and rechallenge. (B) Blood loss during primary challenge and rechallenge. These data demonstrate that both primary and secondary hemostasis are impaired in VWF2N/2N mice in a severed tail tip injury model.
Statistical analysis of the bleeding time and blood loss in mice with various VWF genotypes using TTT bleeding test
| Two-way ANOVA Multiple comparisons of mice with various VWF genotypes | Bleeding time (minutes) | Blood loss (volume) | ||
|---|---|---|---|---|
| Primary (P) | Rechallenge (P) | Primary (P) | Rechallenge (P) | |
| C57BL/6J vs VWF+/+ | .5929 | .5003 | .8962 | .8308 |
| C57BL/6J vs VWF2N/+ | .4209 | .2956 | .1783 | .1879 |
| C57BL/6J vs VWF2N/2N | .0175* | .0107* | .0001*** | .0209* |
| C57BL/6J vs VWF2N/− | .0788 | .6932 | .3813 | .8563 |
| C57BL/6J vs VWF−/− | .0551 | .0011** | .7122 | .0430* |
| C57BL/6J vs VWF+/− | .4009 | .4441 | .7818 | .7962 |
| VWF+/+ vs VWF2N/+ | .1983 | .0999 | .1622 | .1468 |
| VWF+/+ vs VWF2N/2N | .0062** | .0025** | .0002*** | .0172* |
| VWF+/+ vs VWF2N/− | .0302* | .2973 | .3397 | .7008 |
| VWF+/+ vs VWF−/− | .0206* | .0003*** | .6326 | .0345* |
| VWF+/+ vs VWF+/− | .7955 | .9616 | .8960 | .9762 |
| VWF2N/+ vs VWF2N/2N | .1068 | .1185 | .0079** | .3001 |
| VWF2N/+ vs VWF2N/− | .3313 | .5124 | .6324 | .2545 |
| VWF2N/+ vs VWF−/− | .2553 | .0209* | .3251 | .4633 |
| VWF2N/+ vs VWF+/− | .1032 | .0732 | .1064 | .1169 |
| VWF2N/2N vs VWF2N/− | .5124 | .0288* | .0020** | .0322* |
| VWF2N/2N vs VWF−/− | .6260 | .4311 | .0004*** | .7595 |
| VWF2N/2N vs VWF+/− | .0017** | .0012** | <.0001**** | .0108* |
| VWF2N/- vs VWF−/− | .8662 | .0037** | .6108 | .0641 |
| VWF2N/- vs VWF+/− | .0109* | .2481 | .2507 | .6606 |
| VWF−/− vs VWF+/− | .0069** | <.0001**** | .5190 | .0235* |
| Two-way ANOVA Multiple comparisons of mice with various VWF genotypes | Bleeding time (minutes) | Blood loss (volume) | ||
|---|---|---|---|---|
| Primary (P) | Rechallenge (P) | Primary (P) | Rechallenge (P) | |
| C57BL/6J vs VWF+/+ | .5929 | .5003 | .8962 | .8308 |
| C57BL/6J vs VWF2N/+ | .4209 | .2956 | .1783 | .1879 |
| C57BL/6J vs VWF2N/2N | .0175* | .0107* | .0001*** | .0209* |
| C57BL/6J vs VWF2N/− | .0788 | .6932 | .3813 | .8563 |
| C57BL/6J vs VWF−/− | .0551 | .0011** | .7122 | .0430* |
| C57BL/6J vs VWF+/− | .4009 | .4441 | .7818 | .7962 |
| VWF+/+ vs VWF2N/+ | .1983 | .0999 | .1622 | .1468 |
| VWF+/+ vs VWF2N/2N | .0062** | .0025** | .0002*** | .0172* |
| VWF+/+ vs VWF2N/− | .0302* | .2973 | .3397 | .7008 |
| VWF+/+ vs VWF−/− | .0206* | .0003*** | .6326 | .0345* |
| VWF+/+ vs VWF+/− | .7955 | .9616 | .8960 | .9762 |
| VWF2N/+ vs VWF2N/2N | .1068 | .1185 | .0079** | .3001 |
| VWF2N/+ vs VWF2N/− | .3313 | .5124 | .6324 | .2545 |
| VWF2N/+ vs VWF−/− | .2553 | .0209* | .3251 | .4633 |
| VWF2N/+ vs VWF+/− | .1032 | .0732 | .1064 | .1169 |
| VWF2N/2N vs VWF2N/− | .5124 | .0288* | .0020** | .0322* |
| VWF2N/2N vs VWF−/− | .6260 | .4311 | .0004*** | .7595 |
| VWF2N/2N vs VWF+/− | .0017** | .0012** | <.0001**** | .0108* |
| VWF2N/- vs VWF−/− | .8662 | .0037** | .6108 | .0641 |
| VWF2N/- vs VWF+/− | .0109* | .2481 | .2507 | .6606 |
| VWF−/− vs VWF+/− | .0069** | <.0001**** | .5190 | .0235* |
P < .05; **P < .01; ***P < .001; ****P < .0001.
Interestingly, primary blood loss in the VWF2N/2N group was significantly greater than that in the VWF−/− group (263.2 ± 219.8 and 43.4 ± 68.9 μL, respectively), but blood loss from rechallenge was comparable for the 2 groups (155.9 ± 112 and 137.8 ± 112.1 μL, respectively). Blood loss during primary and rechallenge bleeding in the VWF2N/− group (73.4 ± 67.9 and 27.1 ± 43.7 μL, respectively) was significantly less than in the VWF2N/2N group. Bleeding time and blood loss in the VWF2N/− group were comparable to the VWF−/− group in primary bleeding, but VWF2N/− mice bled much less than VWF−/− mice during rechallenge (Figure 7A-B;,Table 2). These data suggest that both primary and secondary hemostasis was impaired in VWF2N/2N mice in a severed tail tip injury model.
Discussion
In this study, we developed the first VWF2N mouse model to simulate a homozygous or compound heterozygous human 2N VWD in mice using CRISPR/Cas 9 strategy. In our model, the 2354G>a [G785E] mutation was introduced into exon 18 of mVWF, resulting in a qualitatively defective VWF that is completely incapable of binding to FVIII but is otherwise normal. Plasma FVIII levels were severely decreased in VWF2N/2N and VWF2N/− mice, comparable to VWF−/− mice. The whole blood CT was prolonged, and thrombin generation was attenuated in VWF2N/2N mice compared with wild-type animals. VWF2N/2N mice exhibited a different bleeding phenotype compared with VWF−/− mice in TVT, TAT, and TTT injury models.
2N VWF mutations within the D′D3 domain FVIII binding site are associated with varying degrees of decreased affinity for FVIII.21,42 When various 2N variants identified in humans were introduced into cloned mVWF cDNA and examined in in vitro studies, they exhibited similar levels of the functional defect in binding to both human and mouse FVIII. mVWF with variant G785E completely lacks the capacity to bind FVIII in 2N VWD model mice. To our knowledge, this is the first 2N VWD mouse model that mimics severe human 2N VWD with normal levels of plasma VWF, but that VWF cannot bind FVIII, resulting in plasma FVIII levels as low as those in VWF-deficient mice.
Low plasma FVIII levels in VWF2N/2N mice are a secondary pathology because 2N VWF cannot act as a carrier for FVIII, protecting FVIII from proteinase degradation. Indeed, when normal VWF is infused into VWF2N/2N mice, endogenous plasma FVIII:C is rescued within 30 minutes after VWF infusion. Even when only 45 ± 23 mU/mL of hVWF remained at 4 hours after infusion, plasma mouse FVIII levels rose to 282 ± 58 mU/mL, suggesting that FVIII is synthesized normally in VWF2N/2N mice and can be sustained in blood circulation if it encounters normal functional VWF with which it associates. Although the normal ratio of VWF and FVIII in functional units in blood is 1:1, our results suggest that 1 mU of VWF has the capacity to protect ∼6 mU of functional FVIII:C in blood. This implies that for the treatment of severe 2N VWD, it may be more effective to provide a consistently low level (eg, 20%) of functional VWF rather than sporadic high-dose VWF treatment.
In humans, individuals with heterozygous VWF2N generally do not exhibit the bleeding phenotype, and their plasma FVIII levels may be normal or only slightly reduced.42,43 In our VWF2N/+ mice, plasma FVIII:C levels were similar to VWF+/+ littermates but significantly higher than VWF+/− mice. We speculate that VWF-dependent FVIII storage in ECs in VWF2N/+ mice might be more effective than in VWF+/− mice because 2N VWF can still associate with FVIII in an intracellular acidic environment.44,45 More bioavailable FVIII may then be secreted from ECs in VWF2N/+ mice and subsequently be stabilized in plasma by normal VWF derived from the VWF+ allele, leading to higher plasma FVIII:C levels than in VWF+/− mice. VWF2N cannot bind FVIII in the neutral pH environment of plasma, so in VWF2N/2N and VWF2N/− mice, FVIII is quickly degraded once secreted from ECs.
Patients with 2N VWD generally exhibit mild to moderate coagulopathy in the clinic and laboratory and may be misdiagnosed as having hemophilia A because both diseases exhibit normal VWF levels and low FVIII:C in plasma. The diagnosis of 2N VWD relies on the FVIII-binding assay to examine the binding capacity of VWF for FVIII. In our VWF2N mouse model, homozygous animals completely lack FVIII-binding capacity. When functional hemostatic properties were assessed by ROTEM, the whole blood CTs in VWF2N/2N and VWF2N/−, but not VWF−/−,, mice were prolonged when compared with wild-type mice. Interestingly, parameters from nWB-TGA showed impairment in VWF2N/2N mice but not VWF−/− mice, suggesting that nWB-TGA could be a valuable assay to assess blood coagulopathy in 2N VWD.
Besides acting as a carrier protein for FVIII in blood circulation, VWF plays an important role in primary hemostasis at sites of injury, functioning as a bridge to mediate platelet adhesion to exposed subendothelial matrix. When we examined bleeding time and blood loss in TVT or TAT injury models, neither VWF2N/2N nor VWF2N/− mice exhibited any hemorrhagic phenotype in either primary or rechallenge bleeding tests. We reasoned that although 2N VWF lacks the binding capacity for FVIII, it can still effectively interact with platelets through GPIbα11 or GPIIb/IIIa12 and exposed collagen13 to initiate platelet adhesion, trigger intracellular signaling to activate platelets,46,47 and promote platelet aggregation to form a clot. VWF−/− mice would lack the platelet effect of VWF, although both have FVIII markedly reduced.
Although VWF is important for primary hemostasis, matrix proteins also can directly bind to platelets (eg, collagen, thrombospondin, and fibronectin).11,48-51 Thus, in VWF−/− mice, platelets could still attach to the injured sites through other adhesive glycan proteins to initiate primary hemostasis although VWF is absent. However, due to lack of VWF to facilitate platelet accumulation and aggregation and absent delivery of FVIII to the clot, secondary hemostasis is impaired, leading to prolonged bleeding and blood loss in VWF−/− mice. Our results from both TVT and TAT models support this mechanism.
We found that VWF2N/2N mice lost a considerable amount of blood in the TTT injury model, in which the tail tip was severed, resulting in a scenario of an amputation wound. Blood loss in VWF2N/2N mice was 19- and 50-fold more than VWF+/+ littermates under the TTT primary and rechallenge bleeding tests, respectively. This was in striking contrast to the results obtained from TVT and TAT injury models, in which both bleeding time and blood loss in the VWF2N/2N group were comparable to wild-type mice. These results suggest that the subendothelial matrix is critical for VWF to initiate primary hemostasis. In the TVT or TAT injury models, 2N VWF could bind to exposed subendothelial matrix collagens and arrest platelets for forming a clot and stopping bleeding, although the clot formed in VWF2N/2N mice might be less stable because plasma FVIII levels are low and thrombin generation is impaired. In the TTT model, the wound is such that the entire vessel needs to occlude to prevent blood loss, and hemostasis cannot rely on simply bridging a transected gap, as in the TVT or TAT injury models.
While the mechanism for more bleeding in VWF2N/2N versus VWF−/− mice during primary bleeding in the TTT injury model is unclear, we speculate that because multiple pathways can participate in primary hemostasis, they may compete or facilitate each other depending on conditions. Indeed, it is known that at least 4 platelet-adhesive glycoproteins (VWF, fibrinogen, fibronectin, and thrombospondin) share a common binding receptor, GPIIbIIIa, on activated platelets,52 and shear rates can impact platelet adhesion to those glycoproteins and collagen.53-60 Among these adhesive glycoproteins, VWF may have a competitive advantage in bridging platelets and collagen because VWF has 2 binding sites per monomer for each.9,61-63 In VWF2N/2N mice, the binding of 2N VWF to platelets or exposed subendothelial matrix components during primary hemostasis may hinder binding of other adhesive proteins to platelets and/or matrix, thereby blocking other potential alternative hemostatic rescue pathways, and in primary severed tail bleeding tests, bleeding was more severe than even in VWF−/− animals.
In VWF−/− mice, there is no VWF to compete, so other platelet-adhesive proteins may have greater access and act as an alternative mechanism between collagen and platelets to initiate hemostasis. Although hemostasis initiated in VWF−/− mice was also impaired and although their FVIII levels were as low as VWF2N/2N mice, the specific molecular entities involved and the functional details may be different, as suggested by our data from ROTEM and nWB-TGA. Further mechanistic studies to explore the hemostatic properties of VWF2N/2N mice are warranted.
In summary, we have developed a novel mouse model by gene editing with both the pathophysiology and clinical phenotype observed in patients with severe type 2N. We show that 2N VWF is dysfunctional in its critical FVIII-carrying function but normal in all other respects. Plasma levels of VWF are normal in VWF2N/2N mice, but this VWF is incapable of binding FVIII. VWF2N/2N mice exhibit impaired hemostatic properties and a severe bleeding phenotype in a severed tail tip injury model. This is a unique model of 2N VWD that can be used to investigate the biological properties of VWF/FVIII association in hemostasis and beyond.
Acknowledgments
This work was supported by the National Heart, Lung, and Blood Institute, National Institutes of Health (grants HL139847, HL081588, HL144457, HL112614 [R.R.M.], and HL102035 [Q.S.]) and support from the Versiti Blood Research Foundation (R.R.M.), the Children’s Hospital of Wisconsin Foundation (Q.S.), and the Midwest Athletes Against Childhood Cancer and Bleeding Disorders (MACC) fund (Q.S.).
Authorship
Contribution: Q.S. designed experiments, analyzed data, and wrote the manuscript; S.A.F. and J.G.M. designed and performed experiments, analyzed data, and edited the manuscript; H.Y., C.P., P.A.M., and J.A.S. performed experiments and analyzed data; J.R. maintained mouse colonies and performed genotyping; H.W. helped to generate type 2N mice and edited the manuscript; and R.R.M. designed and supervised research and helped to write the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Qizhen Shi, Section of Hematology/Oncology, Department of Pediatrics, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: qshi@versiti.org.

