

Key Points
scRNAseq identifies two CD4+ GM-CSF+ T-cell subsets in the colon during GVHD that can be distinguished based on expression of IFN-γ.
CD4+ GM-CSF+ IFN-γ− T cells constitute an IL-7–responsive, transcriptionally and developmentally distinct colitogenic TH GM-CSF + lineage.

Abstract
Gastrointestinal (GI) tract involvement is a major determinant for subsequent morbidity and mortality arising during graft-versus-host disease (GVHD). CD4+ T cells that produce granulocyte-macrophage colony stimulating factor (GM-CSF) have emerged as central mediators of inflammation in this tissue site as GM-CSF serves as a critical cytokine link between the adaptive and innate arms of the immune system. However, cellular heterogeneity within the CD4+ GM-CSF+ T-cell population due to the concurrent production of other inflammatory cytokines has raised questions as to whether these cells have a common ontology or if a unique CD4+ GM-CSF+ subset exists that differs from other defined T helper subtypes. Using single-cell RNA sequencing analysis (scRNAseq), we identified two CD4+ GM-CSF+ T-cell populations that arose during GVHD and were distinguishable according to the presence or absence of interferon-γ (IFN-γ) coexpression. CD4+ GM-CSF+ IFN-γ− T cells, which emerged preferentially in the colon, had a distinct transcriptional profile, used unique gene regulatory networks, and possessed a nonoverlapping T-cell receptor repertoire compared with CD4+ GM-CSF+ IFN-γ+ T cells as well as all other transcriptionally defined CD4+ T-cell populations in the colon. Functionally, this CD4+ GM-CSF+ T-cell population contributed to pathologic damage in the GI tract that was critically dependent on signaling through the interleukin-17 (IL-7) receptor but was independent of type 1 interferon signaling. Thus, these studies help to unravel heterogeneity within CD4+ GM-CSF+ T cells that arise during GVHD and define a developmentally distinct colitogenic T helper subtype GM-CSF+ subset that mediates immunopathology.
Introduction
Granulocyte-macrophage colony stimulating factor (GM-CSF) has emerged as an important mediator of inflammation in many disease states,1-4 including graft-versus-host disease (GVHD) in which GM-CSF has a critical role in promoting pathologic damage in the gastrointestinal (GI) tract.5-8 Although cell types such as B cells, epithelial cells, endothelial cells, and fibroblasts have been documented to produce GM-CSF,9-12 CD4+ T-cell production of GM-CSF seems to be the primary pathway by which inflammation is propagated by this cytokine.13-15 Because GM-CSF is sensed exclusively by cells of the myeloid lineage,16 this cytokine functions as an important bridge linking the innate and adaptive arms of the immune system.17,18 Specifically, the production of GM-CSF can activate dendritic cells, monocytes, polymorphonuclear leukocytes, and macrophages that can then induce downstream inflammatory pathways.19-21 Mechanistically, CD4+ T-cell–derived GM-CSF links innate and adaptive immunity by promoting indirect alloantigen presentation in mesenteric lymph nodes (mLNs),7 which has been shown to be a critical pathway for the propagation of GVHD in the GI tract.22
A frequent observation in human studies and murine inflammatory disease models, including GVHD, has been the identification of heterogeneous populations of CD4+ GM-CSF+ T cells in tissue culture and in vivo within specific target tissues; specifically, those that make GM-CSF alone or cells that produce GM-CSF in conjunction with other T helper–defining cytokines, most often interferon-γ (IFN-γ) or interleukin-17 (IL-17).6,7,23-26 This finding has raised the question as to whether these cells constitute a subset of T helper 1 (Th1) and/or T helper 17 (Th17) cells, or if a distinct T-helper GM-CSF subset exists within this heterogeneous population that differentiates these cells ontologically from more classically defined Th1 and Th17 cells.27 To date, however, characterization of CD4+ GM-CSF+ T-cell heterogeneity within specific tissue sites in inflammatory diseases such as GVHD has not been addressed. The goal of the current study was to define existing heterogeneity within CD4+ GM-CSF+ T cells arising in the GI tract, as well as assess the temporal kinetics, transcriptional profiles, developmental trajectories, and functional roles of these CD4+ T-cell populations.
Material and methods
Mice
C57BL/6 (B6) (H-2b) and Balb/c (H-2d) mice were bred in the Biomedical Resource Center at the Medical College of Wisconsin (MCW) or purchased from The Jackson Laboratory (Bar Harbor, ME). Donor and recipient mice were 5 to 12 weeks of age and were sex matched in all transplantation studies. All mice were housed in the Association for Assessment and Accreditation of Laboratory Animal Care–accredited Biomedical Resource Center of MCW.
Other detailed methods
All other methods are described in the supplemental Material and methods.
Results
Two distinct CD4+ GM-CSF+ T-cell populations emerge in the colon during GVHD
We observed that 2 distinct populations of CD4+ GM-CSF+ T cells emerged in GVHD target organs: those that coexpressed IFN-γ and those that were IFN-γ− (Figure 1A). Notably, the percentage of total CD4+ GM-CSF+ T cells was significantly increased in the colon relative to other tissue sites (Figure 1B). Also, the subset of CD4+ T cells that were GM-CSF+ IFN-γ− represented a significantly larger percentage of CD4+ GM-CSF+ T cells in the colon than either the liver, lung, or spleen (Figure 1C). These cells were not present in the colons of naive mice or in recipients of syngeneic marrow grafts (data not shown) but did develop in animals reconstituted with minor histocompatibility antigen–mismatched grafts, albeit at reduced frequencies (supplemental Figure 1). Examination of other T-cell–derived inflammatory cytokines28 revealed that both subsets produced tumor necrosis factor-α, although a higher percentage of CD4+ GM-CSF+ IFN-γ+ T cells expressed this cytokine, whereas production of IL-6 and IL-17 was relatively modest in the 2 populations (supplemental Figure 2). Time course studies revealed no change in the absolute number of CD4+ T cells in the colon within the first 3 weeks’ posttransplantation (Figure 1D). However, there was a significant increase in the number of CD4+ GM-CSF+ IFN-γ− T cells (Figure 1E), with no difference in CD4+ GM-CSF+ IFN-γ+ T cells over this same interval (Figure 1F).
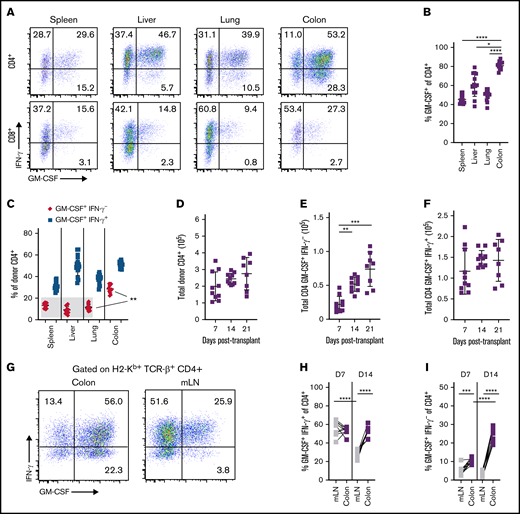
Emergence of distinct CD4+ GM-CSF+ T-cell populations in the GI tract during acute GVHD. Lethally irradiated Balb/c mice were transplanted with B6 bone marrow (5 × 106) plus spleen cells (adjusted to yield an αβ T-cell dose of 8 × 105). (A) Representative dot plots depicting the expression of GM-CSF and IFN-γ in donor-derived (H-2Kb+, TCR-β+) CD4+ (top) or CD8+ (bottom) T cells isolated from indicated tissues 21 days posttransplantation. (B) The percentage of donor CD4+ T cells expressing GM-CSF in each tissue. (C) The percentage of donor-derived CD4+ T cells that were either GM-CSF+ IFN-γ− or GM-CSF+ IFN-γ+ in each tissue. Results from panels B and C are from 2 independent experiments (n = 15). (D-F) The total number of CD4+ T cells (D), CD4+ GM-CSF+ IFN-γ− T cells (E), and CD4+ GM-CSF+ IFN-γ+ T cells (F) in the colon at 7, 14, and 21 days’ posttransplantation. Data are from 2 experiments (n = 8-10 mice). (G) Representative dot plot gated on donor CD4+ T cells expressing GM-CSF and/or IFN-γ in the colon and mLN at 14 days’ posttransplantation. (H-I) The percentage of GM-CSF+ IFN-γ+ (H) and GM-CSF+ IFN-γ− (I) donor CD4+ T cells in the indicated tissues at 7 and 14 days’ posttransplantation. Data for panels H and I are from 2 experiments (n = 9-10 mice per time point). *P < .05, **P < .01, ***P < .001, ****P < .0001. D7, day 7; D14, day 14.
Emergence of distinct CD4+ GM-CSF+ T-cell populations in the GI tract during acute GVHD. Lethally irradiated Balb/c mice were transplanted with B6 bone marrow (5 × 106) plus spleen cells (adjusted to yield an αβ T-cell dose of 8 × 105). (A) Representative dot plots depicting the expression of GM-CSF and IFN-γ in donor-derived (H-2Kb+, TCR-β+) CD4+ (top) or CD8+ (bottom) T cells isolated from indicated tissues 21 days posttransplantation. (B) The percentage of donor CD4+ T cells expressing GM-CSF in each tissue. (C) The percentage of donor-derived CD4+ T cells that were either GM-CSF+ IFN-γ− or GM-CSF+ IFN-γ+ in each tissue. Results from panels B and C are from 2 independent experiments (n = 15). (D-F) The total number of CD4+ T cells (D), CD4+ GM-CSF+ IFN-γ− T cells (E), and CD4+ GM-CSF+ IFN-γ+ T cells (F) in the colon at 7, 14, and 21 days’ posttransplantation. Data are from 2 experiments (n = 8-10 mice). (G) Representative dot plot gated on donor CD4+ T cells expressing GM-CSF and/or IFN-γ in the colon and mLN at 14 days’ posttransplantation. (H-I) The percentage of GM-CSF+ IFN-γ+ (H) and GM-CSF+ IFN-γ− (I) donor CD4+ T cells in the indicated tissues at 7 and 14 days’ posttransplantation. Data for panels H and I are from 2 experiments (n = 9-10 mice per time point). *P < .05, **P < .01, ***P < .001, ****P < .0001. D7, day 7; D14, day 14.
The predilection of CD4+ GM-CSF+ T cells to reside in the colon led us to examine the mLNs and colon at earlier time points as donor T cells are primed in this site before trafficking to the GI tract.22 The percentage of CD4+ GM-CSF+ IFN-γ+ T cells in the mLN comprised a majority of all CD4+ T cells at day 7 before declining to ∼25% by day 14 (Figures 1G-H). Concurrently, there was no difference in the frequency of these cells in the colon at 7 and 14 days (ie, ∼50%), and this finding approximated what was observed at 3 weeks’ posttransplantation (Figure 1C), indicative of a relatively stable population. In contrast, the percentage of CD4+ GM-CSF+ IFN-γ− T cells in the mLN averaged only ∼5% on days 7 and 14, which was substantially lower compared with CD4+ GM-CSF+ IFN-γ+ T cells (Figure 1I). However, whereas there was no appreciable change in the frequency of CD4+ GM-CSF+ IFN-γ+ T cells in the colon, the percentage of CD4+ GM-CSF+ IFN-γ− T cells significantly increased between days 7 and 14. Collectively, these studies showed that CD4+ GM-CSF+ IFN-γ− T cells emerge in the colon during GVHD with different temporal kinetics and without significant prior accumulation in the mLN, suggesting that these two CD4+ GM-CSF subsets represented developmentally distinct populations.
Single-cell RNA sequencing confirms 2 distinct CD4+ GM-CSF+ T-cell subsets within the colonic T-cell population
We next examined whether CD4+ GM-CSF+ IFN-γ+ and CD4+ GM-CSF+ IFN-γ− T cells represented transcriptionally discrete populations by performing single-cell RNA sequencing (scRNAseq) on sorted donor-derived T cells from the colons of GVHD mice. This analysis revealed 7 transcriptionally distinct clusters (Figure 2A) based on an optimal clustering resolution that provided the most meaningful discrimination between CD4+ and CD8+ T-cell subsets (supplemental Figure 3A). Nearly all cells expressed high levels of Cd3e, with little to no contamination from other cell types such as CD14+ macrophages, CD19+ B cells, or NK1.1+ NK cells (supplemental Figure 3B). The number of genes and unique molecular identifiers (UMIs) for all clusters is depicted in supplemental Figure 3C. The small number of cells in cluster 6 expressed lower levels of CD3e and higher levels of Cd14 and H2-Ab1 (MHC-II) and were deemed to be sort contaminants. Compared with CD8+ T cells, CD4+ T cells were a more transcriptionally heterogeneous population (supplemental Figure 3D). A heat map depicting the top 10 genes expressed in each CD4+ T-cell cluster (clusters 2-5) compared with the average expression in all other cells is shown in Figure 2B. Notably, we observed that the clusters of CD4+ T cells could be defined by the expression or absence of IFN-γ (encoded by Ifng) and GM-CSF (encoded by Csf2), namely GM-CSF− IFN-γ+ (cluster 2), GM-CSF+ IFN-γ+ (cluster 3), GM-CSF+ IFN-γ− (cluster 4), and GM-CSF− IFN-γ− (cluster 5) (Figure 2C). There was minimal expression of other T-cell–derived inflammatory cytokines (ie, IL-6, IL-17, tumor necrosis factor-α, IL-22) that play a role in the pathophysiology of GI GVHD.28 Discordance was observed between transcriptional expression of tumor necrosis factor-α and the intracellular detection of this protein, whereas results were largely concordant for IL-6 and IL-17. We also observed no expression of IL-3, which was previously reported in an in vitro–generated population of neuroinflammatory CD4+ GM-CSF+ T cells.24
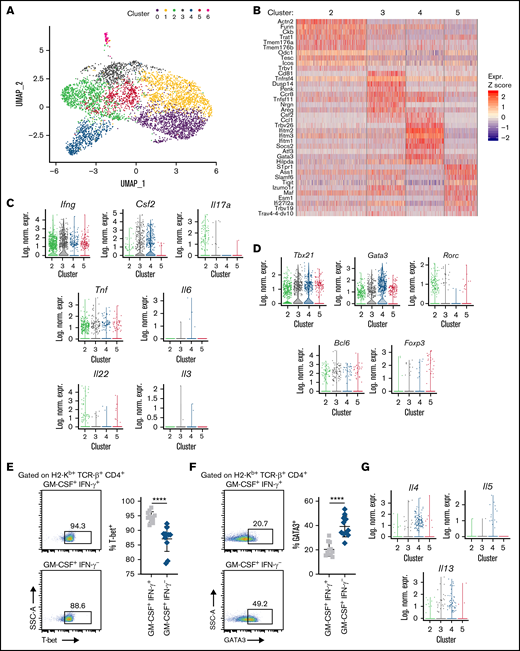
scRNAseq defines 2 transcriptionally discrete CD4+ GM-CSF+ T-cell populations in the colon. Lethally irradiated Balb/c mice were transplanted with B6 bone marrow (5 × 106) plus spleen cells (adjusted to yield an αβ T-cell dose of 8 × 105). (A) Uniform manifold approximation and projection (UMAP) dimensional reduction of scRNAseq data of flow-sorted, donor-derived T cells from the colons of transplant recipients 21 days posttransplantation. Unsupervised clustering using Seurat revealed 7 transcriptionally distinct clusters using a resolution of 0.4. (B) Heat map depicting z-scored expression of the top 10 overexpressed genes in CD4+ T-cell clusters (clusters 2-5) relative to all other cells. (C-D) Violin plots showing log-normalized expression of the indicated proinflammatory cytokines (C) and CD4+ T-cell lineage-defining transcription factors (D). (E-F) Representative flow cytometry dot plots and summary data depicting T-bet (E) and Gata3 (F) expression on donor CD4+ GM-CSF+ IFN-γ+ or CD4+ GM-CSF+ IFN-γ– T cells in the colon. (G) Violin plots showing log-normalized expression of the indicated Th2-associated genes in CD4+ T-cell clusters. Data are from 2 to 3 experiments. In UMAP and violin plots, each dot represents a single cell. ****P < .0001. Log. Norm. Expr., log-normalized expression, SSC-A, side scatter area.
scRNAseq defines 2 transcriptionally discrete CD4+ GM-CSF+ T-cell populations in the colon. Lethally irradiated Balb/c mice were transplanted with B6 bone marrow (5 × 106) plus spleen cells (adjusted to yield an αβ T-cell dose of 8 × 105). (A) Uniform manifold approximation and projection (UMAP) dimensional reduction of scRNAseq data of flow-sorted, donor-derived T cells from the colons of transplant recipients 21 days posttransplantation. Unsupervised clustering using Seurat revealed 7 transcriptionally distinct clusters using a resolution of 0.4. (B) Heat map depicting z-scored expression of the top 10 overexpressed genes in CD4+ T-cell clusters (clusters 2-5) relative to all other cells. (C-D) Violin plots showing log-normalized expression of the indicated proinflammatory cytokines (C) and CD4+ T-cell lineage-defining transcription factors (D). (E-F) Representative flow cytometry dot plots and summary data depicting T-bet (E) and Gata3 (F) expression on donor CD4+ GM-CSF+ IFN-γ+ or CD4+ GM-CSF+ IFN-γ– T cells in the colon. (G) Violin plots showing log-normalized expression of the indicated Th2-associated genes in CD4+ T-cell clusters. Data are from 2 to 3 experiments. In UMAP and violin plots, each dot represents a single cell. ****P < .0001. Log. Norm. Expr., log-normalized expression, SSC-A, side scatter area.
To determine whether the CD4+ T-cell clusters fell into defined T helper lineages, we examined the expression of Thelper 1 (Th1) (T-bet/Tbx21), Th2 (Gata3), Th17 (RORγt/Rorc), T follicular helper (Bcl6), and regulatory T cell (Foxp3) lineage-defining transcription factors. Notably, both T-bet and GATA3 were expressed in each CD4+ GM-CSF+ T-cell population. T-bet expression was similar in clusters 3 and 4, whereas expression of GATA3 was approximately twofold higher in CD4+ GM-CSF+ IFN-γ− T cells (Figure 2D). None of the 4 clusters exhibited significant expression of Rorc, Bcl6, or Foxp3. Intracellular protein staining confirmed increased expression of GATA-3 in CD4+ GM-CSF+ IFN-γ– T cells (Figure 2E), whereas there was decreased expression of T-bet in this population (Figure 2F). There was minimal expression of the Th2-associated genes IL-4 and IL-5 in cluster 4 cells (Figure 2G); however, intracellular flow cytometry of ex vivo–stimulated cells revealed increased production of IL-4 and IL-5 in these cells relative to CD4+ GM-CSF+ IFN-γ+ T cells (supplemental Figure 4). Thus, these data indicated that CD4+ GM-CSF+ T cells segregate into 2 transcriptionally distinct populations based on the presence or absence of IFN-γ expression.
CD4+ GM-CSF+ IFN-γ− T cells have a distinct transcriptional signature compared with CD4+ GM-CSF+ IFN-γ+ T cells
A more detailed transcriptome analysis of these two CD4+ GM-CSF+ T-cell subsets (ie, cluster 3 vs 4) revealed 117 differentially expressed genes, with 37 overexpressed in cluster 4 and 80 overexpressed in cluster 3 based on defined cutoff criteria (|ln (fold change) | >0.5 and padjusted < .0001) (a full list is available in supplemental Table 1), representing ∼2.1% of the total sequenced transcriptome (Figure 3A). CD4+ GM-CSF+ IFN-γ+ T cells were characterized by expression of higher levels of costimulatory genes (Tnfrsf9/CD137, Tnfrsf4/OX40, Cd160, and Cd81) but also augmented expression of the inhibitory receptors Lag3 (3.2-fold) and Pdcd1/programmed cell death protein 1 (PD-1) (2.0-fold) (Figure 3B). Further examination by flow cytometry revealed significantly decreased cell surface expression of PD-1 and LAG3 but not CD81 on CD4+ GM-CSF+ IFN-γ− T cells compared with CD4+ GM-CSF+ IFN-γ+ T cells (Figure 3C). CD69, which is a marker for CD4+ tissue resident memory T cells,29,30 had 1.7-fold higher expression on CD4+ GM-CSF+ IFN-γ− T cells (Figure 3D). There was also a modest but statistically significant increase in surface expression of CD69 (Figure 3E-F). However, there was no transcriptional evidence or surface expression of integrin αɛ (CD103), which is a known binding partner for the integrin β7 and expressed on some CD4+ tissue resident memory populations in the intestinal tract31 (Figure 3D-E). CD4+ GM-CSF+ IFN-γ− T cells were distinguished by increased transcription (Figure 3G) and surface expression (Figure 3H-I) of Il1rl1 (ST2) whose expression on T cells has been associated with pathogenicity in the GI tract during GVHD.32,33 Thus, these data showed that CD4+ GM-CSF+ IFN-γ− T cells possessed a transcriptional signature as well as differential expression of cell surface proteins that was distinct from that of CD4+ GM-CSF+ IFN-γ+ T cells.
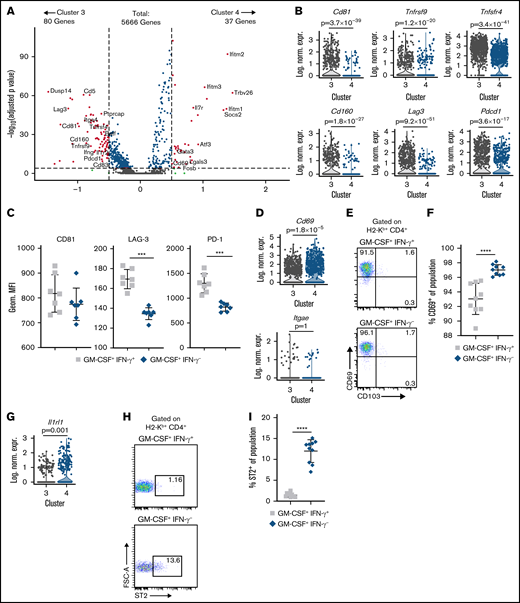
CD4+ GM-CSF+ IFN-γ– T cells possess a distinctive transcriptional signature. (A) Volcano plot showing over/underexpressed genes in CD4+ GM-CSF+ IFN-γ− T cells (cluster 4) vs CD4+ GM-CSF+ IFN-γ+ T cells (cluster 3) from the colons of GVHD mice 21 days’ posttransplantation. Cutoff parameters were |ln(fold change)| > 0.5 and padjusted < .0001. (B) Violin plots showing log-normalized expression of the indicated T-cell activation markers and immune checkpoint inhibitors in clusters 3 and 4. P values are depicted. (C) Summary data of PD-1, Lag3, and CD81 surface expression by flow cytometry on CD4+ GM-CSF+ IFN-γ+ and GM-CSF+ IFN-γ– cells in the colon at 21 days’ posttransplantation. Data are from 2 experiments (n = 7 mice per group). (D) Violin plots showing log-normalized expression of CD69 and CD103 on cells in clusters 3 and 4. (E-F) Representative flow cytometry dot plots (left) and summary data (right) depicting CD69 and CD103 expression on donor CD4+ GM-CSF+ IFN-γ+ or CD4+ GM-CSF+ IFN-γ– T cells in the colon at 21 days posttransplantation. Data are from 2 experiments (n = 9 mice per group). (G) Violin plots showing log-normalized expression of Il1r1 (ST2) in clusters 3 and 4. (H-I) Representative flow cytometry dot plots (left) and summary data (right) depicting ST2 expression on donor CD4+ GM-CSF+ IFN-γ+ or CD4+ GM-CSF+ IFN-γ− T cells in the colon at 21 days’ posttransplantation. Data are from 2 experiments (n = 9 mice per group). In violin plots, each dot represents a single cell. ***P < .001, ****P < .0001. Geom. MFI, geometric mean fluorescence intensity; Log. Norm. Expr., log-normalized expression.
CD4+ GM-CSF+ IFN-γ– T cells possess a distinctive transcriptional signature. (A) Volcano plot showing over/underexpressed genes in CD4+ GM-CSF+ IFN-γ− T cells (cluster 4) vs CD4+ GM-CSF+ IFN-γ+ T cells (cluster 3) from the colons of GVHD mice 21 days’ posttransplantation. Cutoff parameters were |ln(fold change)| > 0.5 and padjusted < .0001. (B) Violin plots showing log-normalized expression of the indicated T-cell activation markers and immune checkpoint inhibitors in clusters 3 and 4. P values are depicted. (C) Summary data of PD-1, Lag3, and CD81 surface expression by flow cytometry on CD4+ GM-CSF+ IFN-γ+ and GM-CSF+ IFN-γ– cells in the colon at 21 days’ posttransplantation. Data are from 2 experiments (n = 7 mice per group). (D) Violin plots showing log-normalized expression of CD69 and CD103 on cells in clusters 3 and 4. (E-F) Representative flow cytometry dot plots (left) and summary data (right) depicting CD69 and CD103 expression on donor CD4+ GM-CSF+ IFN-γ+ or CD4+ GM-CSF+ IFN-γ– T cells in the colon at 21 days posttransplantation. Data are from 2 experiments (n = 9 mice per group). (G) Violin plots showing log-normalized expression of Il1r1 (ST2) in clusters 3 and 4. (H-I) Representative flow cytometry dot plots (left) and summary data (right) depicting ST2 expression on donor CD4+ GM-CSF+ IFN-γ+ or CD4+ GM-CSF+ IFN-γ− T cells in the colon at 21 days’ posttransplantation. Data are from 2 experiments (n = 9 mice per group). In violin plots, each dot represents a single cell. ***P < .001, ****P < .0001. Geom. MFI, geometric mean fluorescence intensity; Log. Norm. Expr., log-normalized expression.
The development of CD4+ GM-CSF+ IFN-γ− T cells is not dependent on signaling through the type 1 interferon receptor
Gene set enrichment analysis revealed that CD4+ GM-CSF+ IFN-γ− T cells had increased expression of IFN-α response genes relative to all other CD4+ T-cell clusters (supplemental Figure 5A). In addition, scRNAseq studies showed that the interferon-induced transmembrane genes Ifitm1, Ifitm2, and Ifitm3 were among the most differentially expressed in cluster 4 vs cluster 3 (supplemental Figure 5B; supplemental Table 1). These genes encode transmembrane proteins that are responsive to type 1 and type 2 interferons and have been shown to protect against select viral infections.34,35 We therefore examined whether type 1 interferon signaling played a role in the emergence of CD4+ GM-CSF+ IFN-γ− T cells by treating mice with either anti-IFNAR1 or an isotype control antibody. IFNAR1 and IFNAR2 subunits constitute a heterodimeric receptor to which all type 1 interferons bind; therefore, blockade of this receptor prevents broad type 1 interferon signaling.36 There was a slight but significant increase in the total number of CD8+ T cells but no difference in overall CD4+ T-cell numbers in the colons of animals treated with anti-IFNAR1 antibody (supplemental Figure 5C). In addition, there was no difference in the frequency of CD4+ T cells that produced either IFN-γ or GM-CSF (supplemental Figure 5D-E) or in the percentage of CD4+ GM-CSF+ IFN-γ− or CD4+ GM-CSF+ IFN-γ+ T cells (supplemental Figure 5F) in anti-IFNAR1 antibody-treated mice. Finally, there was no difference in pathologic damage in the colon between mice treated with anti-IFNAR1 vs an isotype control antibody (supplemental Figure 5G-H), indicating that blockade of type 1 interferon signaling had no effect on the emergence of CD4+ GM-CSF+ IFN-γ− T cells nor did it attenuate pathologic damage.
CD4+ GM-CSF+ IFN-γ− T cells are dependent on signaling through the IL-7R
Further examination of potential pathways that were important for the development of CD4+ GM-CSF+ IFN-γ− T cells revealed that CD4+ GM-CSF+ IFN-γ− T cells also had increased expression of genes involved in the IL-2/STAT5 pathway, which is critical for downstream IL-2 and IL-7 signaling37,38 (Figure 4A). Although there was no difference in expression of Il2ra between clusters 3 and 4, cluster 4 exhibited approximately twofold to threefold higher expression of Il7r (Figure 4B). In addition, there was a modest but significant increase in the surface expression of the IL-7R on CD4+ GM-CSF+ IFN-γ− T cells (Figure 4C-D). To determine whether this population was functionally significant and responsive to IL-7R signaling, recipient animals were treated with either an anti–IL-7Rα or isotype control antibody. Mice that received anti–IL-7Rα antibody had significantly fewer donor CD4+ T cells in the colon 3 weeks posttransplantation, whereas the absolute number of CD8+ T cells was unaffected (Figure 4E). Analysis of cytokine production in donor T cells revealed that IL-7R blockade significantly reduced the proportion of CD4+ T cells that produced GM-CSF, whereas there was a trend toward an increased percentage of CD4+ IFN-γ+ T cells in these mice (Figure 4F-G). Notably, this was due to a specific reduction in the CD4+ GM-CSF+ IFN-γ− T-cell subset, while there was no change in the frequency of CD4+ GM-CSF+ IFN-γ+ T cells and a corresponding increase in the percentage of CD4+ cells that were GM-CSF− IFN-γ+ (Figure 4H). The absolute number of both CD4+ GM-CSF+ IFN-γ− and CD4+ GM-CSF+ IFN-γ+ T cells were significantly reduced in mice treated with anti–IL-7R antibody; however, the reduction was more pronounced in CD4+ GM-CSF+ IFN-γ− T cells (Figure 4I). In contrast, anti–IL-7R antibody administration had no effect on the percentage of CD8+ T cells that secreted GM-CSF and/or IFN-γ (Figure 4J). Histopathologic analysis revealed that animals treated with anti–IL-7R antibody also had significantly less weight loss (supplemental Figure 6A) and pathologic damage in the colon with pathology scores that were no different from those observed in non-GVHD control animals (Figure 4K-L). There was also reduced pathology in the lung and liver of anti–IL-7-treated animals (supplemental Figure 6B). Collectively, these studies revealed that signaling through the IL-7R was critical for the development of CD4+ GM-CSF+ IFN-γ− T cells in the colon, and that blockade of this pathway not only specifically reduced the frequency of this population but also attenuated GVHD-related tissue damage in the GI tract.
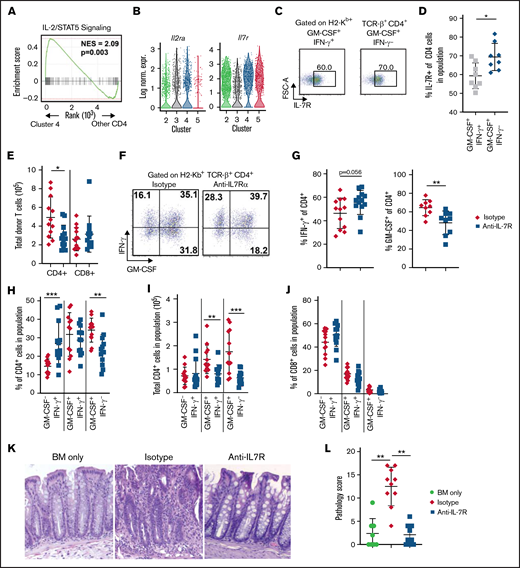
Blockade of IL-7R signaling significantly reduces the emergence of CD4+ GM-CSF+ IFN-γ– T cells and attenuates pathologic damage in the colon during GVHD. (A) Gene set enrichment analysis comparing expression of IL-2/STAT5 signaling-related genes between cluster 4 and other CD4+ T-cell clusters in the scRNAseq data set presented in Figure 2. (B) Violin plots showing log-normalized expression of Il2ra and Il7r in CD4+ T-cell clusters. (C-D) Lethally irradiated Balb/c mice were transplanted with B6 bone marrow (BM) (5 × 106) plus spleen cells (adjusted to yield an αβ T-cell dose of 8 × 105). Representative flow cytometry dot plots (C) and summary data (D) depicting IL-7R expression on donor CD4+ GM-CSF+ IFN-γ+ or CD4+ GM-CSF+ IFN-γ− T cells in the colon at 21 days posttransplantation. Data are from 2 experiments (n = 8 mice per group). (E-L) Lethally irradiated Balb/c mice were transplanted with B6 BM (5 × 106) plus spleen cells (adjusted to yield an αβ T-cell dose of 8 × 105). Recipient mice were treated 3 times weekly intraperitoneally with an anti–IL-7R or isotype control antibody. Colons were harvested 21 days posttransplantation. (E) The total number of donor-derived CD4+ and CD8+ in the colons of mice receiving the indicated treatment. (F) Representative dot plots gated on donor-derived CD4+ cells from the colon showing expression of GM-CSF and IFN-γ. (G) The proportion of donor CD4+ cells isolated from mice in the indicated groups producing IFN-γ (left) and GM-CSF (right). (H-I) The percentage (H) and total number (I) of donor CD4+ T cells isolated from the colon based on expression IFN-γ and/or GM-CSF. (J) The percentage of donor CD8+ T cells isolated from the colon based on expression of IFN-γ and/or GM-CSF. Data in panels E, G, H, I, and J are from 3 experiments (n = 13-14 mice per group). (K) Representative hematoxylin and eosin–stained images of colons 21 days’ posttransplantation. Original magnification is ×100. (L) Pathologic scores of colon tissue from mice transplanted with BM alone or BM and spleen cells and then treated with an isotype control or anti–IL-7R antibody using a semi-quantitative scoring system. Data are from 2 experiments (n = 8-10 mice per group). *P < .05, **P < .01, ***P < .001. FSC-A, forward scatter area; Log Norm. Expr., log-normalized expression; NES, normalized enrichment score.
Blockade of IL-7R signaling significantly reduces the emergence of CD4+ GM-CSF+ IFN-γ– T cells and attenuates pathologic damage in the colon during GVHD. (A) Gene set enrichment analysis comparing expression of IL-2/STAT5 signaling-related genes between cluster 4 and other CD4+ T-cell clusters in the scRNAseq data set presented in Figure 2. (B) Violin plots showing log-normalized expression of Il2ra and Il7r in CD4+ T-cell clusters. (C-D) Lethally irradiated Balb/c mice were transplanted with B6 bone marrow (BM) (5 × 106) plus spleen cells (adjusted to yield an αβ T-cell dose of 8 × 105). Representative flow cytometry dot plots (C) and summary data (D) depicting IL-7R expression on donor CD4+ GM-CSF+ IFN-γ+ or CD4+ GM-CSF+ IFN-γ− T cells in the colon at 21 days posttransplantation. Data are from 2 experiments (n = 8 mice per group). (E-L) Lethally irradiated Balb/c mice were transplanted with B6 BM (5 × 106) plus spleen cells (adjusted to yield an αβ T-cell dose of 8 × 105). Recipient mice were treated 3 times weekly intraperitoneally with an anti–IL-7R or isotype control antibody. Colons were harvested 21 days posttransplantation. (E) The total number of donor-derived CD4+ and CD8+ in the colons of mice receiving the indicated treatment. (F) Representative dot plots gated on donor-derived CD4+ cells from the colon showing expression of GM-CSF and IFN-γ. (G) The proportion of donor CD4+ cells isolated from mice in the indicated groups producing IFN-γ (left) and GM-CSF (right). (H-I) The percentage (H) and total number (I) of donor CD4+ T cells isolated from the colon based on expression IFN-γ and/or GM-CSF. (J) The percentage of donor CD8+ T cells isolated from the colon based on expression of IFN-γ and/or GM-CSF. Data in panels E, G, H, I, and J are from 3 experiments (n = 13-14 mice per group). (K) Representative hematoxylin and eosin–stained images of colons 21 days’ posttransplantation. Original magnification is ×100. (L) Pathologic scores of colon tissue from mice transplanted with BM alone or BM and spleen cells and then treated with an isotype control or anti–IL-7R antibody using a semi-quantitative scoring system. Data are from 2 experiments (n = 8-10 mice per group). *P < .05, **P < .01, ***P < .001. FSC-A, forward scatter area; Log Norm. Expr., log-normalized expression; NES, normalized enrichment score.
CD4+ GM-CSF+ IFN-γ− T cells have highly biased Vβ gene usage and possess a unique T-cell receptor repertoire
To further delineate differences between CD4+ GM-CSF+ IFN-γ− T cells and other CD4+ T-cell clusters, we examined the T-cell receptor (TCR) repertoires of CD4+ T cells within the scRNAseq data set. The distribution of clonotype sizes (ie, number of cells with the same TCR-α and TCR-β CDR3 sequences) revealed that clusters 2, 3, and 5 had similar distributions in which a majority of cells were part of expanded clones (Figure 5A). In contrast, cluster 4 was highly dominated (∼80%) by singletons with very few expanded clones (Figure 5A-B). Examination of shared clonotype sequences revealed significant overlap between clusters 2, 3, and 5, with ∼30% to 40% of all sequences being shared between these 3 clusters (Figure 5C-D). Conversely, there was only minimal overlap between cluster 4 and other CD4+ T-cell populations with only ∼8% shared clonotypes (Figure 5D). We also examined TCR-β variable chain (ie, Vβ) gene usage, which showed that clusters 2, 3, and 5 displayed varied Vβ gene usage, with no specific Vβ gene comprising >21% of the sequenced TCRs (Figure 5E; supplemental Table 2). In contrast, nearly 40% of the TCRs in cluster 4 used Vβ26, whereas Vβ26 represented no more than 3% of Vβ gene usage in all other clusters. Violin plots depicting the most highly used Vβ genes revealed Vβ discordance between clusters 3 and 4, as cluster 3 was more dominated by Vβ13-2, whereas cluster 4 cells predominantly used Vβ26 (Figure 5F). Collectively, these data showed that CD4+ GM-CSF+ IFN-γ− T cells have a highly biased Vβ family usage and possess a TCR repertoire that is distinct from all other colonic CD4+ T cells.
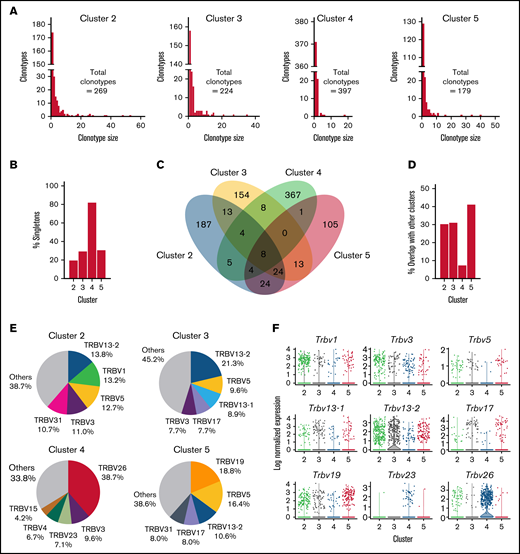
CD4+ GM-CSF+ IFN-γ− T cells possess a unique TCR repertoire. Single cells from the scRNAseq data set presented in Figure 2 were profiled by targeted sequencing of TCR-α and TCR-β chains. A clonotype was defined as one or more cells having an identical CDR3 DNA sequence in both TCR-α and TCR-β genes. (A) The clonotype distribution of each CD4+ T-cell cluster of the scRNAseq data set. Data for each cluster are plotted as the number of individual clonotypes on the y-axis vs the size of each clonotype (number of cells) on the x-axis. (B) The percentage of cells that were singleton clonotypes (clonotype size of one) within each CD4+ T-cell cluster. (C) Venn diagram depicting the overlap of clonotype sequences between CD4+ T-cell clusters. (D) The percentage of overlap in clonotype sequences between the indicated CD4+ T-cell cluster and all other clusters. (E) The proportion of clonotypes that used the indicated Vβ gene in each of the 4 clusters. The top 5 Vβ genes for each cluster are labeled (the full list is provided in supplemental Table 2). (F) Violin plots depicting log-normalized expression of TCR Vβ in cells in the scRNAseq data set. Each dot represents a single cell.
CD4+ GM-CSF+ IFN-γ− T cells possess a unique TCR repertoire. Single cells from the scRNAseq data set presented in Figure 2 were profiled by targeted sequencing of TCR-α and TCR-β chains. A clonotype was defined as one or more cells having an identical CDR3 DNA sequence in both TCR-α and TCR-β genes. (A) The clonotype distribution of each CD4+ T-cell cluster of the scRNAseq data set. Data for each cluster are plotted as the number of individual clonotypes on the y-axis vs the size of each clonotype (number of cells) on the x-axis. (B) The percentage of cells that were singleton clonotypes (clonotype size of one) within each CD4+ T-cell cluster. (C) Venn diagram depicting the overlap of clonotype sequences between CD4+ T-cell clusters. (D) The percentage of overlap in clonotype sequences between the indicated CD4+ T-cell cluster and all other clusters. (E) The proportion of clonotypes that used the indicated Vβ gene in each of the 4 clusters. The top 5 Vβ genes for each cluster are labeled (the full list is provided in supplemental Table 2). (F) Violin plots depicting log-normalized expression of TCR Vβ in cells in the scRNAseq data set. Each dot represents a single cell.
CD4+ GM-CSF+ IFN-γ− T cells possess a discrete developmental trajectory and gene regulatory network
Given the disparities in TCR repertoires between cells in cluster 4 vs all other clusters, we examined cell differentiation relationships using Monocle 2, which employs a machine-learning algorithm to infer cell developmental trajectories from scRNAseq gene expression data.39,40 This algorithm, which predicts trajectories over pseudo time, delineated that cluster 4 was the most developmentally distinct from the other 3 clusters (Figure 6A-B). Monocle further defined 9 different states based on a trajectory-based analysis of the scRNAseq data (Figure 6C). This analysis revealed that cluster 4 cells had a significantly higher percentage of cells in Monocle state 1 with >80% of this population residing in that state (Figure 6D), whereas cluster 3 cells had virtually no cells in that state, indicative of a divergent cellular trajectory.
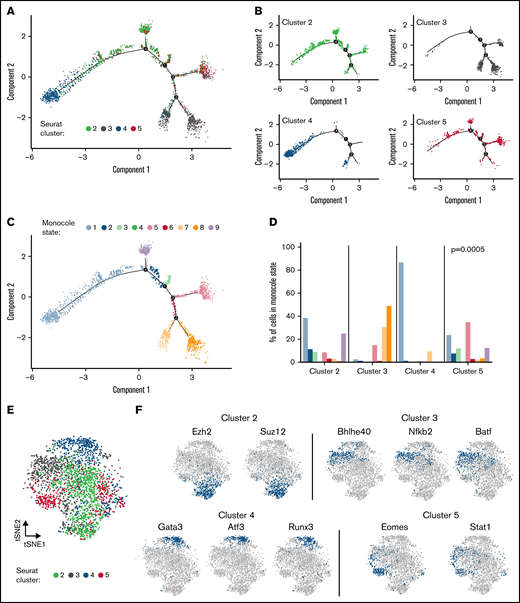
CD4+ GM-CSF+ IFN-γ− T cells have a distinct developmental trajectory and use cluster-specific regulons. Single cells from the scRNAseq data set presented in Figure 2 were analyzed by using Monocle2 and SCENIC statistical packages. (A-B) Monocle single-cell trajectory of CD4+ T cells labeled according to cluster in aggregate (A) and depicted as individual clusters (B). (C) Single-cell trajectory of CD4+ T cells as labeled by Monocle state. (D) The percentage of cells from each cluster of cells falling into a particular Monocle state. The P value from Fisher’s exact test comparing the distribution of Monocle states among clusters is shown. (E) t-distributed stochastic neighbor embedding (tSNE) plot of CD4+ T cells mapped based on SCENIC binary regulon activity and labeled according to Seurat cluster. (F) tSNE plots of selected regulons used in CD4+ T cells from clusters 2 to 5.
CD4+ GM-CSF+ IFN-γ− T cells have a distinct developmental trajectory and use cluster-specific regulons. Single cells from the scRNAseq data set presented in Figure 2 were analyzed by using Monocle2 and SCENIC statistical packages. (A-B) Monocle single-cell trajectory of CD4+ T cells labeled according to cluster in aggregate (A) and depicted as individual clusters (B). (C) Single-cell trajectory of CD4+ T cells as labeled by Monocle state. (D) The percentage of cells from each cluster of cells falling into a particular Monocle state. The P value from Fisher’s exact test comparing the distribution of Monocle states among clusters is shown. (E) t-distributed stochastic neighbor embedding (tSNE) plot of CD4+ T cells mapped based on SCENIC binary regulon activity and labeled according to Seurat cluster. (F) tSNE plots of selected regulons used in CD4+ T cells from clusters 2 to 5.
T-cell differentiation is also driven by the coordinated expression of transcription factors and downstream target genes (ie, regulons), which establish a gene expression profile and can be delineated by using SCENIC (single cell regulatory network inference and clustering), which is a computational method for gene regulatory network reconstruction of scRNAseq data.41,42 Using this approach, we observed that clusters 2 to 5 had distinct regulon activity based on SCENIC binary activity matrices (Figure 6E). A focused examination of cluster 3 vs 4 cells showed enriched regulon activity for Gata3, Atf3, and Runx3 in cluster 4, whereas cluster 3 was dominated by Bhlhe40, Nfkb2, and Batf (Figure 6F). In contrast, cluster 2 was characterized by Ezh2 and Suz12, whereas cluster 5 was predicted to preferentially activate the Eomes and Stat1 regulons. The composite landscape of regulon activity for all 4 clusters, showing both overlapping and non-overlapping regulons, is detailed in supplemental Figure 7. In addition, the dominant regulons identified in cluster 4 (ie, Gata3, Atf3, Runx3) and the corresponding activity or lack thereof of these regulons in the other 3 clusters are depicted (supplemental Figure 8). Thus, CD4+ GM-CSF+ IFN-γ− T cells had a developmentally distinct cellular differentiation trajectory and used gene regulatory networks that distinguished them from all other CD4+ T-cell clusters.
Discussion
The current report examined the pro-inflammatory role of GM-CSF during GVHD by characterizing CD4+ T cells present in the colon to define existing heterogeneity within CD4+ GM-CSF+ T-cell populations. The impetus for this analysis derived from prior studies in transplant and nontransplant settings, which had questioned whether CD4+ GM-CSF+ T cells constitute a distinct CD4+ T-cell lineage or are more accurately subsumed within a broader Th1 or Th17 umbrella.6,7,23-27 The studies herein identified two CD4+ GM-CSF+ T-cell populations that could be clearly discriminated by the presence or absence of IFN-γ coexpression. Notably, these 2 populations had different temporal kinetics as CD4+ GM-CSF+ IFN-γ+ T cells were present in relatively stable numbers during the first 3 weeks, whereas CD4+ GM-CSF+ IFN-γ− T cells progressively increased during this same interval. In addition, CD4+ GM-CSF+ IFN-γ− T cells had minimal prior accumulation in mLNs compared with CD4+ GM-CSF+ IFN-γ+ T cells, indicating that these CD4+ GM-CSF+ T-cell populations did not behave in a similar biological fashion.
Further characterization using scRNAseq analysis revealed that although CD4+ GM-CSF+ IFN-γ+ and CD4+ GM-CSF+ IFN-γ− T cells had near-equivalent expression of T-bet, CD4+ GM-CSF+ IFN-γ− T cells had a greater than twofold increase in GATA3. This was confirmed by intracellular protein staining, which showed significantly increased GATA3 expression and a commensurate reduction in T-bet in this population. There was also a higher frequency of CD4+ GM-CSF+ IFN-γ− T cells that produced IL-4 and IL-5 cells, which was in accord with GATA3 expression. CD4+ T cells that coexpressed GM-CSF and IFN-γ had increased expression of a variety of costimulatory molecules (CD81, CD134, CD137, and CD160) as well as inhibitory receptors (PD-1 and LAG3), which was more characteristic of a terminally exhausted immune phenotype than their CD4+ GM-CSF+ IFN-γ− counterparts. In contrast, CD4+ GM-CSF+ IFN-γ− T cells possessed a gene set signature that was characterized by increased transcriptional and surface expression of Il1rl1 (ST2), which was noteworthy given that ST2-expressing T cells have been shown to induce tissue damage in the GI tract via ligand interactions with IL-33.32,33 These cells also had modestly increased expression of CD69, which inhibits egress from tissue sites.30 Thus, these data are evidence that CD4+ GM-CSF+ IFN-γ− T cells constitute a pathogenic less terminally differentiated population with unique biological attributes.
scRNAseq studies revealed that the Ifitm1, Ifitim2, and Ifitm3 genes were 3 of the top 6 differentially expressed genes in CD4+ GM-CSF+ IFN-γ− T cells compared with their CD4+ GM-CSF+ IFN-γ+ counterparts. The IFITM gene complex in mice encodes a series of 6 transmembrane interferon response genes that are responsive to both type 1 and type 2 interferons and functionally serve to restrict viral entry and replication.34,35 However, these genes have also been shown to play a role in the adaptive immune response,43 in which the absence of Ifitm1-3 genes in T cells results in preferential skewing toward a Th1 phenotype with a reduction in T-bet expression and IFN-γ production.43,44 We therefore reasoned that type 1 interferon signaling that leads to expression of these genes might be important for restraining IFN-γ production and the development of CD4+ GM-CSF+ IFN-γ− T cells. However, we observed no significant difference in the overall percentage of CD4+ GM-CSF+ IFN-γ− T cells in animals that were treated with anti-IFNAR1 antibody. In addition, pathologic damage in the colon was similar in mice that received control vs anti-IFNAR1 antibody, indicating that type 1 interferon signaling did not seem to be critical for the emergence or pathogenicity of CD4+ GM-CSF+ IFN-γ− T cells.
To unveil a potential mechanistic pathway by which CD4+ GM-CSF+ IFN-γ− T cells emerged in the colon and determine whether these cells induced pathogenicity, we focused on the fact that gene set enrichment analysis showed increased expression of genes involved in the IL-2/STAT5 signaling pathway, which is critical for both IL-2 and IL-7 signaling.45 Although there was no difference in expression of the IL-2R on cluster 4 vs cluster 3 cells, scRNAseq analysis revealed differential expression of the IL-7R that was twofold to threefold greater in CD4+ GM-CSF+ IFN-γ− cells as opposed to CD4+ GM-CSF+ IFN-γ+ T cells. The functional significance of this signaling pathway was confirmed by studies which showed that in vivo antibody blockade resulted in a preferential reduction in the frequency of CD4+ GM-CSF+ IFN-γ− T cells, whereas the percentage of CD4+ GM-CSF+ IFN-γ+ T cells was unaffected. Importantly, we also observed that administration of anti–IL-7R antibody reduced pathologic damage in the colon commensurate to the level observed in non-GVHD control animals. The IL-7R is known to be expressed on CD4+ and CD8+ T cells, B cells, and natural killer (NK) cells.37 The latter 2 populations have little role in promoting GVHD within the GI tract,46,47 and we observed no effect of IL-7R signaling blockade on the frequency of CD8+ T cells in this tissue site. Moreover, because GVHD pathology in this model is driven primarily by CD4+ T cells, these data support a causative linkage between this CD4+ GM-CSF+ IFN-γ− T-cell subset and subsequent pathologic damage in the colon, although they do not exclude a role for CD4+ GM-CSF+ IFN-γ+ T cells.
The transcriptional differences between these 2 clusters, coupled with the fact that CD4+ GM-CSF+ IFN-γ+ T cells seemed to be less proliferative, led us to examine individual T-cell repertoires and the extent of clonal expansion. Strikingly, TCR repertoire analysis revealed that ∼40% of all CD4+ GM-CSF+ IFN-γ− T cells in the GI tract used Vβ26, whereas none of the other CD4+ T-cell clusters used >3% of this Vβ gene, indicative of highly biased repertoire usage. In addition, CD4+ T cells in cluster 4 had a disproportionate number of singletons, and the clonotypes within this cluster had virtually no repertoire overlap with TCR sequences observed in any of the other CD4+ T-cell populations. Thus, there seemed to be much less clonal expansion in this CD4+ T-cell cluster, which was consistent with scRNAseq data indicating reduced cell division, and further suggested that CD4+ GM-CSF+ IFN-γ− T cells represented a distinct lineage due to unique TCR sequence usage. To further examine lineage relationships between the four CD4+ T-cell clusters, we used Monocle 2, which employs a machine-learning algorithm to construct cell differentiation trajectories.39 Results from this analysis revealed that the trajectory for CD4+ GM-CSF+ IFN-γ− T cells was clearly distinct from all other CD4+ T-cell populations. In fact, the most divergent trajectories were between cluster 3 and 4, providing further evidence that CD4+ GM-CSF+ IFN-γ− T cells possessed a unique developmental pathway.
To understand differentiation within CD4+ GM-CSF+ T-cell subsets, we employed single-cell transcriptome-derived gene regulatory analysis using SCENIC and identified regulons that distinguished CD4+ GM-CSF+ IFN-γ− T cells from all other CD4+ T-cell populations, including CD4+ GM-CSF+ IFN-γ+ T cells.41,42 Specifically, cluster 4 cells had higher regulon activity of several transcription factors that distinguished these cells from all other clusters, GATA3, Runx3, and ATF3, whereas cluster 3 preferentially used Batf and Bhlhe40, and appeared more analogous to the Batf-dependent CD4+ GM-CSF+ T-cell population described by Ullrich et al.5 Thus, these data would indicate that there are 2 different transcriptional pathways that lead to the emergence of CD4+ GM-CSF+ T cells that are capable of mediating pathologic damage in the GI tract.
GATA3 is the master transcription factor for Th2 differentiation48,49 and leads to the production of the Th2 lineage-defining cytokines IL-4, IL-5, and IL-13.50,51 Notably, however, another primary function of GATA3 is the inhibition of Th1 differentiation and specifically IFN-γ expression.52 In fact, GATA3 has been shown to interfere with the Th1 differentiation program by downregulation of STAT4,52 blocking Runx3-mediated IFN-γ production,53 and silencing Ifng gene expression by epigenetic modifications through the addition of H3K27me3 suppressive marks.54 Thus, we postulate that increased expression of GATA3 may function as a transcriptional rheostat to repress T-bet and Runx3-driven production of IFN-γ. A conjectured role for ATF3 in the differentiation of CD4+ GM-CSF+ IFN-γ− T cells is less apparent given the limited knowledge of how this transcription factor affects T-cell biology. Studies in innate immune cells such as macrophages and NK cells have revealed that ATF3 represses production of IFN-γ,55-57 but this was not observed in T cells, at least in one report.57 Consequently, these limited data engender caution as to how the ATF3 transcriptional regulatory network might affect the development of this CD4+ GM-CSF+ T-cell subset.
In summary, these studies have identified a discrete CD4+ GM-CSF+ IFN-γ− T-cell subset that preferentially emerges in the GI tract during GVHD. These cells are distinguishable from their corresponding CD4+ GM-CSF+ IFN-γ+ T-cell counterparts, as well as all other CD4+ T cells in the colon, by virtue of a distinct transcriptional profile, a nonoverlapping T-cell repertoire, and the use of unique gene regulatory networks. Although CD4+ GM-CSF+ IFN-γ+ T cells were more similar to other CD4+ clusters and had Th1-like features, CD4+ GM-CSF+ IFN-γ− T cells constituted a discrete IL-7–responsive population that was responsible for inducing pathologic damage in the colon. Thus, these results help to unravel heterogeneity within CD4+ GM-CSF+ T cells that arise during GVHD and provide support for the existence of a T helper GM-CSF lineage that differs from other defined T helper subtypes.
Acknowledgments
This research was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (F30 HL143870, C.P.; R01 HL139008 and R01 HL126166, W.R.D.), National Institute of Diabetes and Digestive and Kidney Diseases (F30 DK127526, M.Y.K.), and National Institute of Allergy and Infectious Diseases (R01 AI125741 and R01 AI148403, W.C.). C.P. and M.Y.K. are members of the Medical Scientist Training Program at MCW, which is partially supported by a training grant from the National Institute of General Medical Sciences (T32-GM080202). This research was completed in part with computational resources and technical support provided by the Research Computing Center at MCW.
Authorship
Contribution: C.P. and E.H. conducted and designed experiments and wrote the manuscript; C.Y.-Y., A.K., M.Y.K., and Y.C. performed experiments and analyzed data; J.E. and J.A.M. performed the blinded pathologic analysis; J.G. and W.C. analyzed data and edited the manuscript; and W.R.D. designed experiments, supervised the study, and wrote the manuscript.
Conflict-of-interest disclosure: W.R.D. receives research funding from Sun Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: William R. Drobyski, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: wdrobysk@mcw.edu.

