


TO THE EDITOR:
Serum and glucocorticoid–regulated kinase 1 (SGK1) modulates activity of downstream effectors including inhibition of the transcription factor FOXO3a, previously shown to increase fetal hemoglobin (HbF) expression in erythroid cells.1 We hypothesized that SGK1 inhibition alleviates FOXO3a inhibition and subsequently increases HbF in red blood cells (RBC). We used a potent and selective inhibitor of SGK1, Compound 16y (Comp16y),2 currently in development for cardiomyopathies and some types of cancer. After Comp16y treatment during erythropoietic differentiation of CD34+ cells, we confirmed target engagement through loss of SGK1 activity with reduced phosphorylation of SGK1 at Thr256 and through alleviation of FOXO3a inhibition with decreased phosphorylation of FOXO3a at Ser315. HbF expression increased significantly compared to controls as quantified by western blotting, high-performance liquid chromatography, and flow cytometry. Gene editing to silence FOXO3a abolished HbF induction by Comp16y. Combining Comp16y with hydroxyurea (HU), the standard of care in sickle cell disease (SCD), resulted in an additive increase in HbF expression. Sickle cell donor CD34+ cells treated with Comp16y were protected from sickling under hypoxia compared to controls.
Novel and safe therapeutic approaches to increase HbF expression in RBC are required to provide improved treatments for β-hemoglobinopathies, including SCD.3 SCD is a genetic disorder characterized by sickle shaped RBC under low oxygen tension and subsequent intravascular hemolysis, vascular inflammation, and painful vaso-occlusive crises .4-7 Individuals with SCD who have hereditary persistence of HbF (HPFH) express ∼30% HbF in their RBC and are generally asymptomatic and free of hemolytic anemia. Indeed, increasing expression of HbF (α2γ2) provides benefit through replacement of βS-globin by γ-globin and dilution of HbS (α2βS2) levels in circulating RBC. Additionally, the presence of residual Gln87 in HbF terminates polymerization of the growing deoxyhemoglobin HbS polymer in an oxygen independent manner even in complete anoxia.8 Increasing HbF level in differentiating erythrocytes prevents cell death under hypoxic conditions found in bone marrow to reduce ineffective erythropoiesis and anemia in SCD.9
Current SCD treatments show limited efficacy or have safety concerns. The standard of care is the ribonucleotide reductase inhibitor HU, which is also used to treat chronic myeloid leukemia and ovarian and skin cancers. In SCD, treatment with HU increases expression of HbF,3-5 but does not achieve levels of HbF as reached in hereditary persistence of HbF, and the rate of nonresponders to HU is high. However, it is important to note that even modest increases in HbF achieved by HU3 results in a decreased incidence of vaso-occlusive crises5 and may have a significant impact on organ damage in SCD as evidenced by a recent study showing that early continuous HU therapy prevents diffuse myocardial fibrosis.10 Voxelotor, a recent therapeutic agent approved for SCD, binds and increases the oxygen affinity of HbS but cannot decrease HbS polymerization under complete anoxia. Both in silico modeling and in vitro assay data indicate that treatments to increase HbF in SCD may be superior to Voxelotor treatment.11 Additional safe mechanisms to increase HbF are required to treat SCD.
SGK1, a downstream serine/threonine kinase in the phosphoinositide-3 kinase cascade, controls multiple physiological processes including cell growth, proliferation, migration, and apoptosis.12,13 SGK1 modulates activity of ion channels (ENaC), Na+-Cl- cotransporters (NCC), membrane transporters, cellular enzymes (GSK3B) and transcription factors (FOXO3a, β-catenin, NF-κB, and SP1).12,14,15 Phosphorylation of SGK1 at Ser315 mediates survival signals in human embryonic kidney cells by inhibiting FOXO3a.14,16 Metformin was shown to induce HbF in erythroid cells through a FOXO3a activation mechanism.1 In small trials, metformin showed a trend to a modest increases in HbF in patients with SCD17,18 and in a large retrospective study lower rates of vaso-occlusive crises and acute chest syndrome were found in patients with SCD who take metformin than in those who do not, however subset analysis failed to show a difference before and after metformin treatment.19
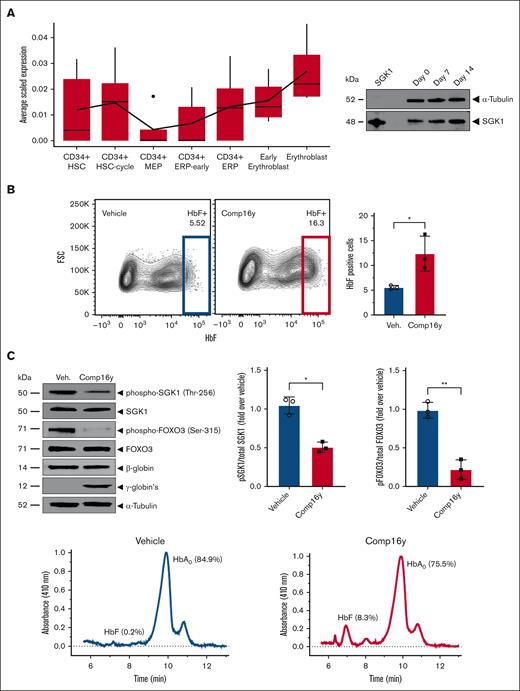
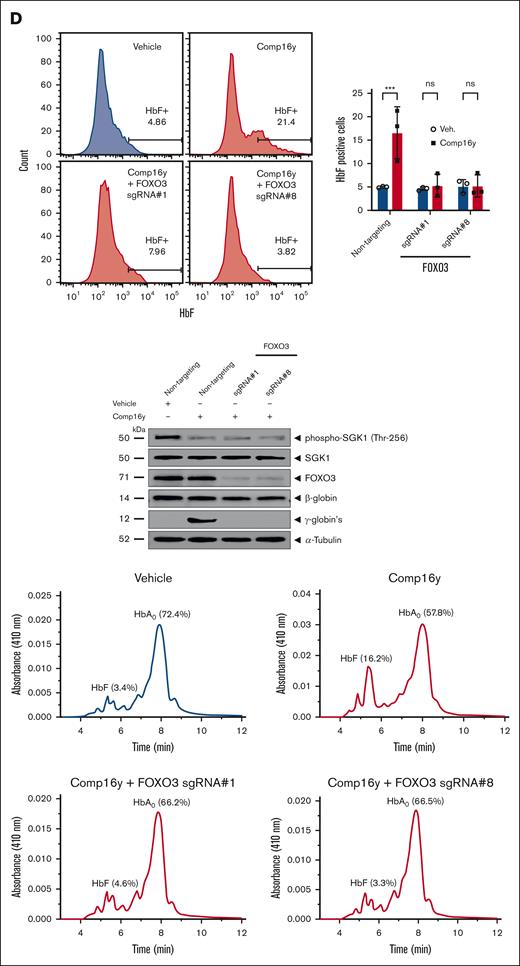
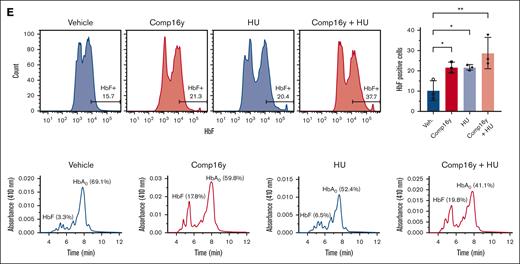
We confirmed expression of SGK1 in CD34+ cells by small-cell RNA sequencing analysis from human erythroid cells showing that SGK1 expression increases during erythroid differentiation, as confirmed by western blotting (Figure 1A). To evaluate the effect of SGK1 inhibition on HbF induction, we treated CD34+ from healthy donors with Comp16y (supplemental Figure 1).2 Quantification of messenger RNA expression revealed an increase in γ-globin after 14-day differentiation, and flow cytometry, western blot, and high-performance liquid chromatography analysis confirmed the induction of HbF (Figure 1B-C; supplemental Figures 2-3). Target engagement and SGK1 inhibition was confirmed by a decrease in SGK1 phosphorylation at Thr256 (Figure 1C), and subsequent decrease in phosphorylation of FOXO3a at Ser315, which relieves inhibition of FOXO3a activity. HbF induction was abolished in FOXO3a knockout cells (Figure 1D; supplemental Figures 2-3), confirming the dependence of HbF induction by Comp16y on FOXO3a. We measured the effect of combining SGK1 inhibition by Comp16y and treatment with HU on HbF induction. SGK1 inhibition alone increases flow cytometric quantification of HbF-positive cells to a similar extent to treatment with HU alone. Interestingly the combination of both further increases the fraction of HbF-positive cells (Figure 1E; supplemental Figures 2-3), suggesting that 2 independent pathways modulate HbF. Whole blood was collected from adult (aged >18 years) sickle cell donors at steady state who had not received a blood transfusion within 3 months in a heparin vacutainer tube under a Western IRB approved protocol (WIRB #1138926) for Sanofi, after obtaining informed consent before the drawing of blood or under a Boston Medical Center IRB approved protocol.
SGK1 inhibition induces fetal hemoglobin via FOXO3 in primary erythroid cells from healthy donors. Inhibition of SGK1 with Comp16y blocks SGK1 activation and phosphorylation of FOXO3a, increases expression of HbF in differentiated human CD34+ erythroid cells, and is additive with HU for HbF induction. Healthy donor CD34+ cells were differentiated in culture as described previously20 in order to confirm Comp16y target engagement assessed by SGK1 and FOXO3a phosphorylation and to measure HbF protein levels after inhibition of SGK1. (A) SGK1 gene expression across human bone marrow single-cells (left) and western blot analysis of SGK1 protein during CD34+ cell differentiation (right) both confirm expression of SGK1. Expression of SGK1 was quantified in 101 935 single bone marrow cells from 8 different donors from the Human Cell Atlas as described in supplemental Methods. The distribution of the average expression of SGK1 across these 8 donors in hematopoietic stem cell (HSC) or defined erythroid cell subsets (MEP, myeloid-erythroid progenitors, ERP, erythroid progenitors) is shown. Western blot is representative of differentiating human CD34+ cells isolated from healthy donor peripheral blood mononuclear cell. (B) SGK1 was inhibited in differentiating healthy donor CD34+ cells by Comp16y (5 μM), a selective and potent inhibitor of SGK1,2 leading to an increase in HbF-positive cells (F-cells) by day 14. Panels are representative flow cytometry plots from a single donor (left). Comp16y significantly increases HbF-positive cells compared to vehicle control (0.1% dimethyl sulfoxide) (2.2-fold change) quantified by flow cytometry (right) as described previously.21 Two-tailed t test, ∗P < .05 (N = 3). (C) Representative western blot of 14-day differentiated healthy donor CD34+ cells treated with Comp16y (5 μM) (left) shows that inhibition of SGK1 alleviates FOXO3a inhibition based on decreased phosphorylation of FOXO3a at Ser315 (4.4-fold decrease, 2-tailed t test, ∗∗P < .01, N = 3), increases expression of HbF based on increased Aγ-globin and Gγ-globin and confirms decreased activity of SGK1 based on decreased SGK1 phosphorylation at Thr256 relative to vehicle (2-fold decrease, 2-tailed t test, ∗P < .05, N = 3). Similar results obtained for 3 independent donors. Increased protein expression of HbF (8.3% vs 0.2% of total globin) after Comp16y treatment of differentiating CD34+ cells is confirmed by cation-exchange high-performance liquid chromatography (right) as described in supplemental Methods. (D) HbF induction by Comp16y (5 μM) is abolished in FOXO3a knockout CD34+ cells measured by flow cytometry at day 14 of differentiation. FOXO3a was silenced by CRISPR-Cas9 gene editing using guided-RNA-FOXO3a. Representative flow cytometry plots from a single donor (left) and combined results from 3 donors confirms SGK1 inhibition significantly increases HbF level only in cells treated with nontargeting RNA control. Two-tailed t test, ∗∗∗P < .001, ns = not significant (N = 3). FOXO3a protein silencing, SGK1 inhibition and lack of HbF induction are confirmed by western blot (middle). High-performance liquid chromatography analysis confirmed FOXO3a dependent HbF induction by Comp16y (right). (E) Combining Comp16y with HU results in higher level of expression of HbF compared to Comp16y or HU alone. Panels on the right are representative flow cytometry plots from a single donor. SGK1 inhibition by Comp16y (5 μM) increases flow cytometric quantification of HbF-positive cells in differentiating CD34+ cells to a similar extent as treatment with HU alone (30 μM) (2-fold increase) and combining Comp16y (5 μM) and HU (30 μM) further increases HbF-positive cells by day 14 (3-fold increase), which is confirmed by high-performance liquid chromatography. Two-tailed t test, ∗∗P < .01, ∗P < .05 (N=3).
Next, we evaluated the effect of SGK1 inhibition during erythrocyte differentiation on sickling of the mature RBC. We differentiated and treated CD34+ cells from SCD donors with Comp16y and applied hypoxic conditions to the resultant RBC (2% O2). After confirming HbF induction (Figure 2A) by Comp16y, we measured sickling of fully differentiated SCD CD34+ cells and quantified through imaging flow cytometry as described previously.21 Comp16y treated CD34+ cells are protected from sickling to attain low levels of sickled cells similar to levels observed under normoxia (20% O2) (Figure 2B). Importantly, SGK1 inhibition does not affect erythroid differentiation as shown by normal expression of erythroid differentiation markers CD235a (Figure 2C), Band-3, LRF, ALAS2, and GATA-1 (Figure 2D) with a minor effect on erythroid cell enucleation (Figure 2E). Finally, Giemsa staining of both the healthy and SCD donor CD34+ cells showed equivalent cell morphology after 14-day differentiation after treatment with Comp16y compared to untreated cells (supplemental Figure 4).

SGK1 inhibition induces fetal hemoglobin in CD34+ cells from sickle cell donors and protects from sickling. Comp16y increases HbF and prevents sickling in differentiated CD34+ cells from sickle cell donors. Treatment with Comp16y increases HbF-positive erythroid cells differentiated from SCD donor blood peripheral blood mononuclear cell and treated cells resist sickling triggered by hypoxia (2% oxygen). (A) Treatment with Comp16y (5 μM) increases HbF-positive cells by day 14 of differentiation in representative flow cytometry plots from a single donor (left) and quantification of data from 3 independent sickle cell donors confirm a significant increase in HbF-positive cells (right) (3.5-fold increase). Two-tailed t test, ∗P < .05 (N=3). (B) Sickling under normoxia (20% oxygen) or hypoxia (2% oxygen) was assessed with fully differentiated CD34+ cells from SCD donor PBMC with and without SGK1 inhibition. Representative microscopic images of 21-day differentiated SCD donor CD34+ cells under hypoxia following differentiation in the presence of Comp16y (5 μM) or 0.1% dimethyl sulfoxide vehicle control (left) shows decreased sickling following SGK1 inhibition (2.1-fold decrease) (scale bar is 20 μm). Arrows indicate abnormal shaped cells. Similar results obtained for 3 independent donors. Sickle cell imaging flow cytometry assays were performed using the Amnis ImageStream and cell shape modification was quantitated using IDEAS software to quantify the percentage of sickling cells (middle) as described previously.21 Representative images by sickle cell imaging flow cytometry assays (right) of cell populations under normoxia gated as under normoxia (nonsickled) or hypoxia (2% oxygen) gated as sickled (60× magnification). Mean ± standard error of the mean (N = 3 independent HbSS SCD donors) and ∗P < .05 (two-tailed t test). (C) Flow cytometric analysis of CD235a+ cells on day 21 indicates normal differentiation of CD34+ cells treated with Comp16y (5 μM). Panels on left are representative flow cytometry plots from a single donor and quantification of data from 3 independent sickle cell donors show no significant decrease in CD235+ cells (middle). Two-tailed t test, ns = not significant (N = 3). Western blot analysis and quantification of selected erythroid differentiation markers (Band-3, LRF, ALAS2, GATA-1) on day 14 are all similar between Comp16y and vehicle treated SCD CD34+ cells when normalized to α-tubulin, confirming normal erythroid cell differentiation. A representative western blot (right top) and statistical analysis of all data (right bottom) are shown. Two-tailed t test, ns = not significant, N = 3. (D) Enucleation observed for fully differentiated SCD CD34+ cells on day 21 for both Comp16y and vehicle treatments shows a minor but significant effect of SGK1 inhibition on enucleation. This minor decrease in enucleation is consistent with normal erythroid differentiation. Two-tailed t test, ∗∗P < .01, N = 3.
SGK1 inhibition induces fetal hemoglobin in CD34+ cells from sickle cell donors and protects from sickling. Comp16y increases HbF and prevents sickling in differentiated CD34+ cells from sickle cell donors. Treatment with Comp16y increases HbF-positive erythroid cells differentiated from SCD donor blood peripheral blood mononuclear cell and treated cells resist sickling triggered by hypoxia (2% oxygen). (A) Treatment with Comp16y (5 μM) increases HbF-positive cells by day 14 of differentiation in representative flow cytometry plots from a single donor (left) and quantification of data from 3 independent sickle cell donors confirm a significant increase in HbF-positive cells (right) (3.5-fold increase). Two-tailed t test, ∗P < .05 (N=3). (B) Sickling under normoxia (20% oxygen) or hypoxia (2% oxygen) was assessed with fully differentiated CD34+ cells from SCD donor PBMC with and without SGK1 inhibition. Representative microscopic images of 21-day differentiated SCD donor CD34+ cells under hypoxia following differentiation in the presence of Comp16y (5 μM) or 0.1% dimethyl sulfoxide vehicle control (left) shows decreased sickling following SGK1 inhibition (2.1-fold decrease) (scale bar is 20 μm). Arrows indicate abnormal shaped cells. Similar results obtained for 3 independent donors. Sickle cell imaging flow cytometry assays were performed using the Amnis ImageStream and cell shape modification was quantitated using IDEAS software to quantify the percentage of sickling cells (middle) as described previously.21 Representative images by sickle cell imaging flow cytometry assays (right) of cell populations under normoxia gated as under normoxia (nonsickled) or hypoxia (2% oxygen) gated as sickled (60× magnification). Mean ± standard error of the mean (N = 3 independent HbSS SCD donors) and ∗P < .05 (two-tailed t test). (C) Flow cytometric analysis of CD235a+ cells on day 21 indicates normal differentiation of CD34+ cells treated with Comp16y (5 μM). Panels on left are representative flow cytometry plots from a single donor and quantification of data from 3 independent sickle cell donors show no significant decrease in CD235+ cells (middle). Two-tailed t test, ns = not significant (N = 3). Western blot analysis and quantification of selected erythroid differentiation markers (Band-3, LRF, ALAS2, GATA-1) on day 14 are all similar between Comp16y and vehicle treated SCD CD34+ cells when normalized to α-tubulin, confirming normal erythroid cell differentiation. A representative western blot (right top) and statistical analysis of all data (right bottom) are shown. Two-tailed t test, ns = not significant, N = 3. (D) Enucleation observed for fully differentiated SCD CD34+ cells on day 21 for both Comp16y and vehicle treatments shows a minor but significant effect of SGK1 inhibition on enucleation. This minor decrease in enucleation is consistent with normal erythroid differentiation. Two-tailed t test, ∗∗P < .01, N = 3.
Previous studies revealed that SGK1 inhibition alleviates inflammation,22,23 and SCD is characterized by exacerbated vascular inflammation. SGK1 inhibition could provide added benefit by alleviating inflammation in SCD, and in this study we found that SGK1 inhibition in human macrophages increases polarization to anti-inflammatory M2 macrophages (supplemental Figure 5) and decreases release of proinflammatory cytokines, IL-6 and IL-12p70 (supplemental Figure 6). As reported previously, chronic hemolysis in SCD polarizes macrophages toward a proinflammatory M1 phenotype,24 and chronic hypoxia triggers SGK1 dependent proinflammatory M1 macrophage activation in mice.25 Therefore, these observations indicate a potential additional beneficial effect of SGK1 inhibition by modulation of inflammation and the immune response in SCD.
However, it is important to consider potential safety concerns for chronic SGK1 inhibition. It has been reported that IL-17 production is impaired in TH17 helper T cells from SGK1–/– mice, and in patients with SCD with leg ulcers, elevated IL-17 correlates with a decreased incidence of acute chest syndrome, which is attributed to more effective TH17 helper T-cells clearance of microbial pathogens.26 Thus, a strategy to inhibit SGK1 in SCD could be limited by negative modulation of the immune response and alleviation of the beneficial effects of TH17 helper T cells in patients with SCD. In addition, as indicated by its name, SGK1 expression in epithelial cells is regulated by glucocorticoids. SGK1 and the related kinases SGK2 and SGK3/CISK stimulate ion channel activities, including the voltage-gated K+ channel Kv1.3, Na+, K+-ATPase, KCNE, and ENaC. Importantly, although SGK1-/- mice show a mild phenotype, they have elevated levels of plasma aldosterone and are unable to maintain normal salt balance when restricted to a low Na+ diet.15
To the best of our knowledge, we show for the first time that inhibition of SGK1 by Comp16y activates FOXO3a to increase HbF in human CD34+ cells from healthy and sickle cell donors and protects differentiated SCD erythroid cells from sickling under hypoxia. We demonstrate that HbF increase is dependent on FOXO3a and show a cumulative effect on γ-globin expression after treatment with Comp16y and HU in combination to increase the percentage of HbF-positive cells. Together these studies establish SGK1 inhibition as a potential new therapeutic strategy in SCD. Successful HU therapy in SCD increases in HbF from baseline levels by ∼3% to ∼7%3 even under optimized individualized dosing,27 and limited clinical data with metformin indicates that similar increase in HbF levels above baseline may be possible.18 However, it is not possible to predict the extent of increase in HbF level achievable by SGK1 inhibition and FOXO3a activation in the clinic. Validation of SGK1 as a safe and efficacious therapeutic target for SCD requires extensive in vivo testing to understand whether chronic inhibition of the ubiquitous SGK1 kinase both increases RBC HbF level and decreases disease markers without unwanted side effects. Comp16y and analogs are currently under investigation for long QT syndrome and cancer indications and no undesired effects are observed in preclinical animal studies.
Acknowledgments: The authors thank their colleagues at Sanofi, including Kim Alving and William Kuhlman for helping in HPLC analysis, Dinesh Bangari and Katie Malley for helping in the Giemsa stain images acquisition, Shannon McGrath, Janina Schwarte and Mark Stottlemyer for the flow cytometry ImageStream data generation, Christophe Lechauve and Mithilesh Kumar Jha (0000-0002-9526-0053) for the critical suggestions in the manuscript. Visual abstract was created with BioRender.com.
This study was funded by Sanofi.
Contributions: Y.H. designed and performed experiments, and analyzed data; V.T.L. designed and performed experiments, and analyzed data; N.H. designed and synthesized Comp16y; K.M. and J.A.R. assisted with resources, provided expertise and feedback, and revised the manuscript; S.L. analyzed SGK1 expression in bone marrow cells, provided feedback, and revised the manuscript; D.R.L. and A.H. supervised the study and provided feedback; and D.R.L., Y.H., and A.H. wrote and revised the manuscript.
Conflict-of-interest disclosure: Y.H., N.H., S.L., A.H., and D.R.L. are employees of Sanofi and had equity ownership with Sanofi. The remaining authors declare no competing financial interests.
Correspondence: Yannis Hara, Immunology and Inflammation, Sanofi, 350 Water St, Cambridge, MA 02141; e-mail: yannis.hara@sanofi.com.

