

Key Points
RUNX1 governs the direct differentiation potential of MK-HSCs into MKs.
RUNX1 binds to and regulates MK developmental genes in MK-HSCs.

Abstract
As a transcription factor in the RUNT domain core-binding factor family, RUNX1 is crucial in multiple stages of hematopoiesis, and its mutation can cause familial platelet disorder with a predisposition to acute myeloid leukemia. Previous work has established that RUNX1 is involved in the maturation of megakaryocytes (MKs) and the production of platelets. Recent studies have shown that there exists a subpopulation of hematopoietic stem cells (HSCs) with relatively high expression of von Willebrand factor and CD41 at the apex of the HSC hierarchy, termed MK-HSCs, which can give rise to MKs without going through the traditional differentiation trajectory from HSC via MPP (multipotent progenitors) and MEP (megakaryocyte–erythroid progenitor). Here, by using Runx1F/FMx1-Cre mouse model, we discovered that the MK-HSC to MK direct differentiation can occur within 1 cell division, and RUNX1 is an important regulator in the process. Runx1 knockout results in a drastic decrease in platelet counts and a severe defect in the differentiation from MK-HSCs to MKs. Single cell RNA sequencing (RNAseq) analysis shows that MK-HSCs have a distinct gene expression signature compared with non-MK–HSCs, and Runx1 deletion alters the platelet and MK-related gene expression in MK-HSCs. Furthermore, bulk RNAseq and Cut&Run analyses show that RUNX1 binds to multiple essential MK or platelet developmental genes, such as Spi1, Selp, and Itga2b and regulates their expressions in MK-HSCs. Thus, by modulating the expression of MK-related genes, RUNX1 governs the direct differentiation from MK-HSCs to MKs and platelets.
Introduction
Hematopoietic stem cells (HSCs) consist of heterogeneous subpopulations of cells, and recent studies have revealed a subpopulation of HSCs at the top of the hematopoiesis hierarchy, which has a megakaryocyte (MK) differentiation potential to support platelet production while maintaining a self-renewal capability to give rise to lymphoid-biased HSCs and multipotent progenitors.1,2 Phenotypically, these HSCs are characterized by the expressions of von Willebrand factor (VWF) and CD41 and account for ∼60% of the total HSCs.1 They are MK-biased (termed as MK-HSCs), and 1 study has shown that the HSC clones of MK-biased output are responsible for up to 50% to 60% of all MK progenies.3 The MK-HSCs can contribute directly to the MK-lineage differentiation and express a distinct gene signature associated with increased quiescence and self-renewal.3 However, in-depth investigations of the molecular mechanism and functional regulation of the MK-HSCs remain limited.
RUNX1 is a transcription factor in the RUNT domain core-binding factor family and is indispensable for the establishment of definitive hematopoiesis in vertebrates.4 In human, heterozygous RUNX1 germ line mutations are closely associated with familial platelet disorder, with propensity to develop acute myeloid leukemia (FPD/AML), a rare autosomal dominant disorder characterized by thrombocytopenia, platelet dysfunction, and a markedly elevated risk for myelodysplastic syndrome and leukemia.5-12 In mice, loss-of-function Runx1 mutation causes defects in lymphocyte and MK development and leads to alterations in hematopoietic stem and progenitor cells that include an increased number of committed erythroid or myeloid progenitors and an expansion of the lineage-negative (L–) Sca1+c-Kit+ cell population in the bone marrow (BM).13-17Runx1 deficient MKs have hypolobulated nuclei, underdeveloped cytoplasm, and low DNA ploidy, whereas Runx1–/– HSCs lose quiescence and increase their numbers.18-21 Although the role of RUNX1 and the putative molecular targets of RUNX1 in HSCs have been extensively studied, how RUNX1 is involved in the various subpopulations of HSCs for their differentiation potential remains unclear, particularly given the technical difficulties in studying RUNX1 molecular interactions.9,14,15,17,19,22-32
In this study, by using the Runx1F/FMx1-Cre inducible knockout mouse model, we have examined the role of Runx1 in MK-HSC direct differentiation into MKs and attempted to define the molecular circuitry involved. We found that MK-HSCs can give rise to MKs within 1 cell division under the stimulation by stem cell factor (SCF) and thrombopoietin (TPO) and that Runx1 deficiency impairs such a differentiation capability. RUNX1 can directly bind to genes required for MK development, including Itga2b, Selp, and Pdgf1b, and regulates their expressions in MK-HSCs. Our study identifies RUNX1 as an essential regulator for the direct differentiation of MK-HSC into MKs.
Materials and methods
Mice
The colonies of Mx1-Cre–Runx1F/F mice were generated and maintained as described previously.13,14,33 Runx1F/F and Mx1-Cre–Runx1F/F mice were injected with polyinosinic:polycytidylic acid (pI:pC; GE HealthCare) 4 times at 15 mg/kg in 2-day intervals to obtain the Runx1F/F (control) and Runx1–/– genotypes. BM cells were harvested at 4 weeks after the final injection. All work done on animals was in accordance with the protocols from the institutional animal care and use committee at Cincinnati Children’s Hospital Medical Center.
Single cell culture
For single cell culture, sorted CD41– or CD41+ HSCs were seeded into a 60-well Terasaki plate (Greiner 653102) with 15 μl StemSpan serum-free expansion media (STEMCELL #09600) plus TPO (50 ng/mL) and SCF (50 ng/mL) per well, and cells were plated as single cells into each well. Triplicates were cultured in a humidified 37°C, 5% CO2 incubator. Image was captured at 40 hours culture time, followed by paired daughter cell selection and RNA extraction. RNA was prepared using the SMART-Seq Single Cell PLUS Kit (Takara Bio Inc) followed by a library preparation using SMART-Seq Library Prep Kit (Takara Bio Inc).
10x Genomics scRNAseq analyses
cKit+, Sca-1+, and Lin− (LSK) CD150+ cells were sorted from Runx1f/f and Runx1–/– mice and were processed with Gene Expression Core at Cincinnati Children’s Hospital Medical Center,using 10x Genomics chromium instrument. For both sets of the droplet-based 10x Genomics data, the Cell Ranger Single-Cell toolkit (version 3.1.0), provided by 10x Genomics, was applied to aligning reads and generating the gene-cell unique molecular identifier matrix, using the mm10 as a reference genome. Then, we applied the R package Seurat34 (version 4.0.5) for downstream analyses. Cells with <500 detected genes and genes expressed in <3 cells were removed. Further quality control was applied to remove cells that had total unique molecular identifier counts >7000 and <100. Moreover, cells with a high detection rate (10%) of mitochondrial gene expression were also removed. After quality control, normalization and log transformation were done with a scale factor of 10 000. Subsequently, we selected the features of the top 2000 highly variable genes as the set features. To compare the gene expression under the 2 conditions, we integrated the single cell RNA sequencing (scRNAseq) data and removed their batch effect based on the protocol of Seurat version 3.35 The principal component analysis (PCA) was performed on the integrated scRNAseq data set to reduce the dimension, and the number of selected principal components (PCs) was 30. We further applied the function “FindClusters” to identify cell clusters with the resolution parameter 2. By comparing the expression of cell-type–specific marker genes, we merged the cell clusters and finally achieved the main cell subpopulations. We applied UMAP36 to illustrate the 2-dimensional embedding of high dimension scRNAseq data. Next, we identified the differentially expressed genes for each cell subpopulation using the Wilcoxon rank-sum test and Bonferroni correction. Finally, after identifying the differentially expressed genes under different conditions, we applied R package clusterProfiler37 (version 4.2.2) to perform gene ontology analysis.
CUT&RUN assay
The CUT&RUN experiments were performed as previously described.38 In brief, sorted CD41+ or CD41− HSCs from Runx1f/f and Runx1–/– mice were attached to the concanavalin A–coated magnetic beads and stained with primary anti-RUNX1 antibodies (provided by Yoram Groner20,39) overnight at 4°C. Protein A-micrococcal nuclease (pA-MN) binding was performed at a final concentration of 700ng/ml and targeted digestion with 100 mM CaCl2 on a 0°C block. The released DNA was extracted using a Qiagen MinElute kit and libraries were prepared with the NEBNext Ultra II DNA Library Prep Kit for Illumina (#E7645). Barcoded libraries were sequenced using a HiSeq 2500 sequencer at the DNA core of Cincinnati Children’s Hospital Medical Center.
Statistical analysis
Statistical analyses were done using GraphPad Prism software. All data are shown as mean ± standard error of the mean. Two tailed Student t test was used to determine the statistical significance. Significance was set as ∗P < .05, ∗∗P < .01, and ∗∗∗P < .001.
Additional methods on histology, flow cytometry, western blotting, colony formation, and RNAseq analyses can be found in the supplemental Materials.
Results
Runx1–/– mice suffer thrombocytopenia and defects in MK differentiation
Recent studies have established that a functional subset of HSCs can predominately differentiate into MKs without going through the hierarchy of multipotent progenitors (MPPs) and megakaryocyte-erythrocyte progenitors (MEPs).1 CD41, an important MK marker, is expressed in HSCs, and 90% of the MK-HSCs express CD41, which can efficiently separate those MK-HSCs from the total HSCs.40 Here, we examined the role of RUNX1 in directing the differentiation of CD41+ MK-HSCs to MKs by using the previously reported Runx1F/FMx1-Cre mouse model.13,14,33 The mice were subjected to 4 pI:pC inductions in 2 day intervals, effectively removing exon 4 of Runx1 and rendering an inactive RUNX1 mutant (Figures 1A-C). The pI:pC injections caused a transient inflammatory response, which was mostly resolved after 8 days.41 Peripheral blood counts at various time points after pI:pC injections show thrombocytopenia upon Runx1 deletion accompanied by a mild decrease in white blood cells (Figure 1D), indicating a reliance on RUNX1 for platelet production. We examined the BM cells via flow cytometry, using the CD41 marker in combination with the long-term HSC (LT-HSC) markers CD34-Flt3−CD150+ (supplemental Figure 1A). We detected an increase in the proportion as well as an increase in cell number of CD41+MK-HSCs in the LT-HSC population after Runx1 deletion, suggesting a possible blockage of MK-HSCs during differentiation (Figure 1E; supplemental Figure 2A-C). We also found that an increase in cell numbers in phenotypic granulocyte-monocyte progenitor, MEP, and multipotent common myeloid progenitors in Runx1–/– mice (supplemental Figure 2D,F), likely because of a higher expression of the myeloid progenitor cell surface marker c-Kit, caused by RUNX1 deficiency. We further applied another well-defined flow cytometry method to detect the changes of erythroid-restricted precursor in the BM using markers CD41, CD150, and Endoglin42 (supplemental Figure 1B). We observed an accumulation in MK progenitors, pre-granulocyte/monocyte (pre-GM), pre-megakaryocyte erythrocyte (pre-MegaE), and pre-CFU-E, and a decrease in (colony-forming unit-erythroid (CFU-E) and erythrocyte (Pro-Ery) in Runx1–/– mice, suggesting that RUNX1 deficiency reduces differentiation from bipotent progenitor MEP to erythroid progenitor,but does not suppress MK progenitor generation (supplemental Figure 2E,G). Consistently, hematoxylin and eosin staining shows an abnormal morphology of MKs in the BM and a decreased number of MKs with CD61 staining, consistent with the low platelets number in the peripheral blood (Figure 1F; supplemental Figure 3A).43 Thus, Runx1 knockout in mice causes a defect in MK differentiation and thrombocytopenia among other blood differentiation defects, consistent with previous reports.14,19,21

HSCs and CD41+ MK-HSCs are dysregulated in Runx1–/– mice and accompanied by thrombocytopenia. (A) Schematics of the mouse model of Runx1F/F;Mx1-Cre and Runx1 inducible deletion. (B) Western blotting of the knockout of RUNX1 protein in duplicate mice. (C) Genotyping of exon 4 deletion of Runx1 in Runx1F/F:Mx1-Cre mice. Triplet samples of Runx1F/F:Mx1-Cre (–/–) and Runx1F/F (F/F) are shown. (D) Peripheral blood counts of Runx1F/F;Mx1-Cre mice after pI:pC inductions. Statistics was performed using t test at 4 weeks post deletion between Runx1−/− and Runx1F/F mice. The x-axis indicates the weeks after the final pI:pC injection. (E) Quantification of flow cytometry analysis of MK-HSCs (LSKCD34−Flt3−CD150+CD41+) in the BM at 4 weeks after pI:pC injection. The y-axis is the percentage out of their parental population. (F) hematoxylin and eosin and anti-CD61 (a MK marker) staining of BM from Runx1−/− and Runx1F/F mice 4 weeks post pI:pC injections. The red arrows indicate MK cells. ∗∗∗P < .001 and ∗∗P < .01. ns, not significant.
HSCs and CD41+ MK-HSCs are dysregulated in Runx1–/– mice and accompanied by thrombocytopenia. (A) Schematics of the mouse model of Runx1F/F;Mx1-Cre and Runx1 inducible deletion. (B) Western blotting of the knockout of RUNX1 protein in duplicate mice. (C) Genotyping of exon 4 deletion of Runx1 in Runx1F/F:Mx1-Cre mice. Triplet samples of Runx1F/F:Mx1-Cre (–/–) and Runx1F/F (F/F) are shown. (D) Peripheral blood counts of Runx1F/F;Mx1-Cre mice after pI:pC inductions. Statistics was performed using t test at 4 weeks post deletion between Runx1−/− and Runx1F/F mice. The x-axis indicates the weeks after the final pI:pC injection. (E) Quantification of flow cytometry analysis of MK-HSCs (LSKCD34−Flt3−CD150+CD41+) in the BM at 4 weeks after pI:pC injection. The y-axis is the percentage out of their parental population. (F) hematoxylin and eosin and anti-CD61 (a MK marker) staining of BM from Runx1−/− and Runx1F/F mice 4 weeks post pI:pC injections. The red arrows indicate MK cells. ∗∗∗P < .001 and ∗∗P < .01. ns, not significant.
Runx1 deletion affects direct differentiation from MK-HSCs to MKs
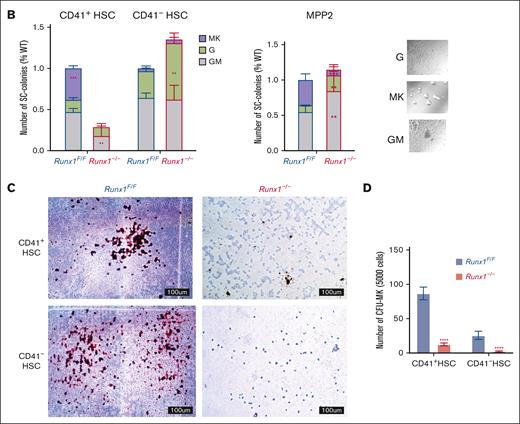
We focused on the MK-HSC subpopulation, which is at the top of hematopoiesis hierarchy. We separated the LSK CD41+ CD34-Flt3−CD150+ MK-HSCs as well as the non-MK–HSCs (LSK CD41−CD34−Flt3−CD150+) via flow cytometry (supplemental Figure 1A). To examine the direct differentiation of these HSCs into MK progenies, we carried out a liquid culture for a short time in the presence of SCF and TPO. We observed a drastic difference in the 2 subpopulations and RUNX1 cell size and shape within 2 days. The Runx1–/– MK-HSCs had almost no large cells of MK-like morphology compared with Runx1F/F MK-HSCs, which gave rise to many cells of MK-like morphology with polyploidy within this time window (Figure 2A; supplemental Figure 3B). To examine the potential of individual MK-HSCs in differentiation, we sorted single MK-HSCs and non-MK–HSCs for a 7-day methycellulose based culture. During the 7-day period we clearly identified MK colonies from granulocytes and GM colonies (Figure 2B, right panel). A quantification of the colony morphologies and numbers in these 2 HSC populations, CD41+ MK-HSCs and CD41– non-MK–HSCs, found that the MK colonies drastically decreased, whereas the granulocyte and GM colonies were maintained in non-MK–HSCs with Runx1 deficiency (Figure 2B, left panel). This points to an interference with direct MK differentiation because of Runx1 deficiency, indicating that RUNX1 is essential in direct megakaryopoesis from MK-HSCs. Because of the methycellulose culture condition, we could only observe the final stage of the MK differentiation based on the morphology of the colonies. To further characterize the colonies, we next examined the differentiation of MK-HSCs in a collagen-based culture condition and quantified the resulting MK-CFUs stained with acetylcholinesterase. A drastically decreased number of MK-CFUs and more immature colonies from the Runx1−/− MK-HSCs were observed (Figure 2C,D). These cellular assays show that Runx1 deficiency causes a loss of the direct MK differentiation capability of MK-HSCs.
RUNX1 deletion affects direct differentiation from MK-HSCs to MKs. (A) Sorted CD41+HSCs (LSKCD34−Flt3−CD150+CD41+), CD41−HSCs (LSKCD34−Flt3−CD150+CD41−) and MPP2 (LSK CD34+Flt3−CD150+CD48+) were plated in StemSpan medium containing SCF and TPO cytokines. Cell images were taken after 2-day culture. Red circles indicate large MK-like cells in the liquid culture dish, and the cell size were measured by cell cytometer. Cells larger than 20um were indicated with green boxes. (B) Quantification of CFU assay from sorted CD41+HSC, CD41−HSC and MPP2. Cells were sorted as single cell into 96 well plates with methylcellulose medium containing multiple cytokines (SCF, TPO, IL-3, IL-6, EPO, IL11, FLT3L, and GM-CSF). After a 7-day culture, colonies were counted based on morphologies. MK, granulocyte, and granulocyte/monocyte colonies were quantified for Runx1–/– and Runx1F/F group 7 days after plating. The y-axis indicates the number of colonies after normalization with Runx1F/F group. (bottom right) Representative views of the MK, G and GM colonies. (C) Sorted CD41+HSC, CD41−HSC cells were cultured in collagen-based semi-solid culture, and after a 7-day culture, MK-CFUs were stained with acetylcholinesterase. Pictures show representative views of the MK-CFUs in CD41+HSC, CD41−HSCs for Runx1–/– and Runx1F/F group. (D) Quantification of the MK-CFUs stained with AChE. ∗∗∗P < .001, ∗∗P < .01, and ∗∗∗∗ P < .0001.
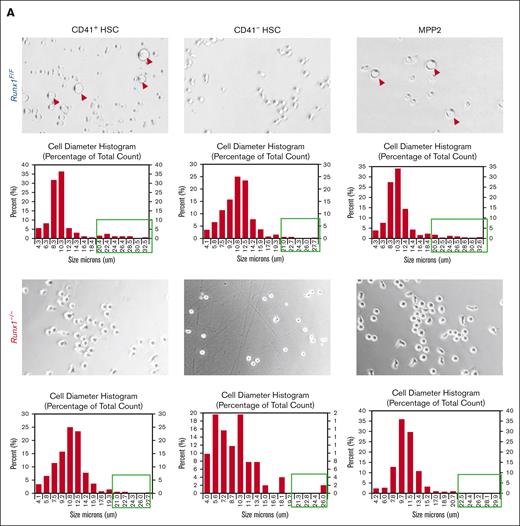
RUNX1 deficiency abolishes MK-HSC direct differentiation into MK within 1 cell division
Because MK-HSCs can give rise to MKs directly, we next examined RUNX1 proficient and deficient HSCs at a single-cell resolution. For single cells cultured in a Terasaki plate, we found that single MK-HSCs from wild-type mice give rise to MK-like large cells within 1 cell division (∼5% frequency), whereas non-MK–HSCs fail to produce any MK-like large cells in their offspring (Figure 3A-C). The frequency of MK-like large daughter cells drastically decreases for MK-HSCs derived from Runx1–/–mice (∼1%) (Figure 3D). This observation raises possibilities that MK-HSCs may give rise to daughter MK cells within 1 cell division and that RUNX1 is important for such a process. To confirm the identity of the paired daughter cells, we performed scRNAseq of those divided large MK-like and small non-MK–like daughter cells from both genotypes. Of the 98 daughter cells that passed quality control measures and yielded an average of 30 million uniquely mappable paired end reads per single cell, 80 were analyzed via differentially expressed gene (DEG) for both large daughter cells and small daughter cells picked from both Runx1F/F and Runx1–/– genotypes. The results show that the MK-like large daughter cells contain a clear MK signature, whereas those smaller non-MK–like daughter cells, either Runx1F/F or Runx1–/–, do not express the MK signature genes and have a relatively higher expression of HSC gene sets (Figure 3E,F; supplemental Figure 4A,B). To further confirm that the isolated MK-HSCs retain multipotent differentiation potential, we also applied a high-throughput single-cell fate potential assay to test the differentiation ability of single MK-HSCs.44 RUNX1 deficiency does not appear to affect MK-HSC lineage differentiation ability into erythroid, granulocytic monocytic, and basophilic lineages (supplemental Figure 5). Taken together, these data further strengthen a regulatory role of RUNX1 in MK-HSC direct differentiation into MKs.

MK-HSCs can directly differentiate to MK within 1 cell division. (A) 60-well Terasaki plate sorted with single cells of CD41+HSC, CD41−HSCs from Runx1–/–and Runx1F/F mice after a 2-day culture in Stemspan medium containing SCF and TPO. (B) Kinetics of the plated single cell divisions. The x-axis indicates the hours after plating the cells post sorting. y-axis indicates the percentage of divided cells in total plated cells. (C) Representative images of the divided large MK-like and small non-MK–like daughter cells. (D) Quantification of the cultured cells as single large MK-like cells, single small non-MK-like cells, double large MK-like cells, and double small non-MK–like cells. (E) Heatmap of key platelet or MK-related gene expressions in large MK-like cells and small non-MK–like cells for Runx1–/–group. (F) Heatmap of key platelet/MK-related gene expressions in large MK-like cells and small non-MK–like cells for Runx1–/–group. ∗∗P < .01 and ∗P < .05.
MK-HSCs can directly differentiate to MK within 1 cell division. (A) 60-well Terasaki plate sorted with single cells of CD41+HSC, CD41−HSCs from Runx1–/–and Runx1F/F mice after a 2-day culture in Stemspan medium containing SCF and TPO. (B) Kinetics of the plated single cell divisions. The x-axis indicates the hours after plating the cells post sorting. y-axis indicates the percentage of divided cells in total plated cells. (C) Representative images of the divided large MK-like and small non-MK–like daughter cells. (D) Quantification of the cultured cells as single large MK-like cells, single small non-MK-like cells, double large MK-like cells, and double small non-MK–like cells. (E) Heatmap of key platelet or MK-related gene expressions in large MK-like cells and small non-MK–like cells for Runx1–/–group. (F) Heatmap of key platelet/MK-related gene expressions in large MK-like cells and small non-MK–like cells for Runx1–/–group. ∗∗P < .01 and ∗P < .05.
scRNAseq reveals a distinct gene signature in MK-HSCs that is altered because of RUNX1 deficiency
To further explore molecular changes in MK-HSCs upon Runx1 deletion, we isolated LSK CD150+ cells from both Runx1f/f and Runx1–/– mouse BM to perform 10x Genomics scRNAseq. To the 8469 captured cells from Runx1f/f genotype and the 10 195 cells from Runx1–/– genotype, we applied gene signatures that combined the reported HSC gene set (MolO)45 and a set of MK genes to cluster the cells into 4 populations: MK-HSCs, non-MK–HSCs, MPP2, and erythroid primed progenitors (Figure 4A,B). MK-HSCs and non-MK–HSCs are distinct, with MK-HSCs having a higher expression of vWF and Itga2b as well as several myeloid lineage–associated genes (eg, Gfi1b, Itga2b, and Pbx1) and MK-lineage associated genes (eg, Gp9, Gp5, Pf4, and Rap1b) (Figure 4C,E; supplemental Figure 6A). We next applied the same clustering to Runx1–/– cells and saw the same 4 populations with distinct gene expressions (Figure 4C, right panel). Particularly, the MK-HSCs showed differential expression patterns of MK regulatory genes in Runx1–/– vs Runx1F/F genotypes, indicating that RUNX1 deficiency dysregulates such gene expression in MK-HSCs (Figure 4D). Consistent with the flow cytometry data in Figure 1E, the number of MK-HSCs in Runx1–/– mice increased compared with that in Runx1f/f mice (Figure 4B), suggesting an accumulation of MK-HSCs possibly related to the dysregulated gene expressions in key regulatory pathways that affect MK differentiation.

scRNAseq reveals a distinct gene signature of MK-HSCs which is altered by Runx1 deficiency. (A) 10x Genomics scRNAseq was used to identify 4 distinct clusters in sorted LSK CD150+ HSCs from Runx1–/–and Runx1F/F group by Seurat program. (B) A quantification of the percentage of cells in the 4 clusters of Runx1F/F and Runx1–/–cells. (C) Heatmap of the DEGs in the 4 clusters of Runx1F/F and Runx1–/–cells. MK-HSCs has a distinct MK-biased signature, and both MK-HSCs and non-MK–HSCs have long-term HSCs signature. (D) Heatmap of the DEGs in the MK-HSCs of Runx1F/F vs Runx1–/–cells. RUNX1 deficiency alters platelet activation related gene expressions in this subset of HSCs, including Pf4, vWF, and Gp5. (E) Feature plot of marker gene Itga2b (CD41) and Gp5 (Glycoprotein V Platelet) for Runx1–/–and Runx1F/F groups.
scRNAseq reveals a distinct gene signature of MK-HSCs which is altered by Runx1 deficiency. (A) 10x Genomics scRNAseq was used to identify 4 distinct clusters in sorted LSK CD150+ HSCs from Runx1–/–and Runx1F/F group by Seurat program. (B) A quantification of the percentage of cells in the 4 clusters of Runx1F/F and Runx1–/–cells. (C) Heatmap of the DEGs in the 4 clusters of Runx1F/F and Runx1–/–cells. MK-HSCs has a distinct MK-biased signature, and both MK-HSCs and non-MK–HSCs have long-term HSCs signature. (D) Heatmap of the DEGs in the MK-HSCs of Runx1F/F vs Runx1–/–cells. RUNX1 deficiency alters platelet activation related gene expressions in this subset of HSCs, including Pf4, vWF, and Gp5. (E) Feature plot of marker gene Itga2b (CD41) and Gp5 (Glycoprotein V Platelet) for Runx1–/–and Runx1F/F groups.
RUNX1 binds to and regulates MK regulatory genes in MK-HSCs
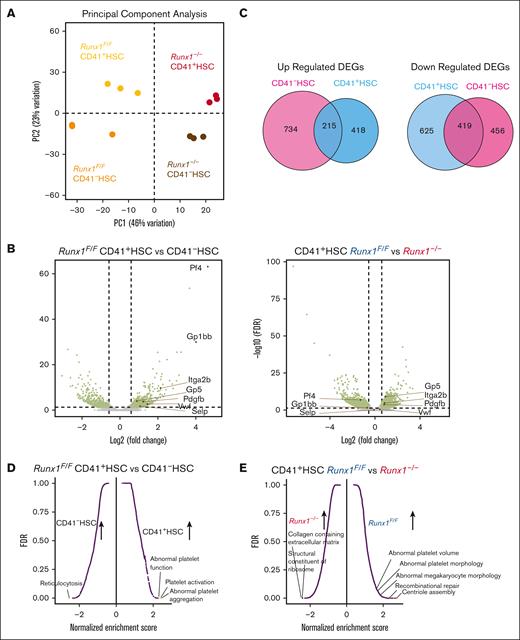
To elucidate the underlying mechanism of altered MK-HSC differentiation by Runx1 deletion, considering that the sequencing depth in 10x Genomics may not be adequate to reveal a full picture of gene regulation in HSCs, we next sorted MK-HSCs and non-MK–HSCs from the Runx1f/f and Runx1–/– genotypes and performed deeper sequencing on bulk RNAs with a NovaSeq SP flow cell. We first performed PCAs of the samples and saw a clear separation of Runx1f/f CD41+, Runx1f/f CD41–, Runx1–/– CD41+, and Runx1–/– CD41– HSCs while confirming the replicates of each genotype samples (Figure 5A,B; supplemental Figure 7A,B). We found 1677 DEGs on comparing Runx1f/f with Runx1–/– CD41+ HSCs. We detected 2375 DEGs on comparing Runx1f/f with Runx1–/– CD41- HSCs. To examine the common DEGs attributed to RUNX1 loss, we overlapped the upregulated and downregulated DEGs between the CD41+ HSC and CD41– HSC sets of samples and found among the 633 upregulated DEGs in CD41+ HSCs, 215 were overlapped with CD41– HSC upregulated DEGs, and among the 1044 downregulated DEGs in CD41+ HSCs, 419 were overlapped with CD41− HSC downregulated DEGs (Figures 5C), indicating that RUNX1 has distinct regulatory roles in the HSC subpopulation. To identify the MK-primed nature of CD41+ HSCs, we further compared the gene expression in Runx1f/f CD41+ HSCs and Runx1f/f CD41– HSCs. When we evaluated the expression level of key factors in MK development via a MK-HSC specific gene signature, we detected increased Itga2b and VWF levels, which are essential markers in MK-HSCs and play a vital role in MK development, in Runx1f/f and Runx1–/– CD41+ HSCs vs CD41– HSCs (Figure 5B; supplemental Figure 7B). Further gene sets enrichment assay (GSEA) pathway enrichment analyses with the overlapping DEGs46 showed that MK differentiation and platelet activation pathways are upregulated drastically in Runx1f/f CD41+ HSCs compared with that in CD41– HSCs (Figure 5D,F; supplemental Figure 7C). These results suggest that CD41+ MK-HSCs are biased or primed toward MK differentiation. A similar MK-primed signature was also seen in Runx1–/– CD41+ HSCs compared with that in CD41– HSCs (supplemental Figure 7D).
RUNX1 regulates platelet and MK regulatory gene expression. (A) PCA plot of RNAseq replicates for each genotype of sorted HSCs. (B) Volcano plot of differential expressed genes in CD41+HSC vs CD41-HSCs from Runx1F/F mice and differential expressed genes in Runx1F/F vs Runx1–/–cells in CD41+HSC. (C) Overlaps of differential expressed genes between CD41+HSC vs CD41–HSCs for Runx1–/–and Runx1F/F genotypes show the commonly changed gene expressions in the cell populations. (D) GSEA analysis of CD41+HSC vs CD41–HSCs in Runx1F/F genotype shows major upregulated pathways in CD41+HSCs including multiple megakaryocyte/platelet related pathways. (E) GSEA analyses of CD41+HSCs for Runx1F/F and Runx1–/–groups show upregulated pathways in WT including platelet activity related pathways. (F) Heatmap of key platelet related gene expressions in CD41+HSC vs CD41−HSCs for Runx1F/F genotype indicates a distinct expression pattern and general increase of platelet related genes in CD41+HSCs. (G) Heatmap of key platelet regulatory gene expression in CD41+HSCs for Runx1F/F and Runx1–/–genotypes shows that RUNX1 deletion alters the gene expression pattern of many platelets-regulatory genes.
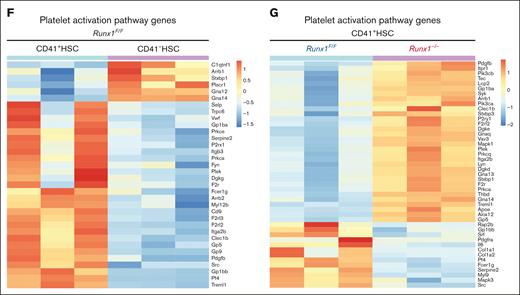
To uncover the functional role of RUNX1 in the MK-primed HSCs, we compared the expression levels of the same set of MK development factors in Runx1f/f and Runx1–/– MK-HSCs. Some, such as Pf4 and Selp, were downregulated, whereas others, such as Itga2b and Pdgfb, were upregulated (Figure 5B,G). Pathway enrichment caused by DEGs revealed dysregulated pathways in MK-HSCs because of RUNX1 deficiency, including multiple DNA repair pathways, cell cycle and cell stress pathways, transforming growth factor β (TGF-β) pathway, and platelet activation pathways (Figure 5E; supplemental Figure 7E). In comparison, in CD41– HSCs, pathway enrichment caused by DEGs showed dysregulated pathways, including TP53 signaling, DNA repair pathways, and inflammatory signaling such as interferon gamma pathways and TGF-β pathway (supplemental Figure 7F). The overall results indicate that RUNX1 regulates key pathways related to platelet activation and signaling in MK-HSCs.
To further examine potential targets of RUNX1, we isolated CD41+ and CD41– HSCs in both genotypes and performed RUNX1 CUT&RUN assay. After alignment and background control using Runx1–/– HSCs (Figure 6A), the CUT&RUN peaks were called with sparse enrichment analysis for CUT&RUN (SEACR) stringent mode, showing that RUNX1 bound regions were mainly localized at intron, transcriptional start sites (TSS), intergenic, and upstream regions (Figure 6C). MOTIF calling analysis revealed RUNX1 specific motifs in both CD41+ and CD41– HSCs; however, the binding selectivity seems to be context dependent, for example, in CD41+ HSCs, RUNX1 enriches the Ets1, Runx1, and Fli1 gene motifs, whereas in CD41– HSCs, RUNX1 enriches the Sox4, Sox7, and Ets1 motifs (Figure 6B).
Platelet and MK regulatory genes in MK-HSCs as possible RUNX1 targets. (A) RUNX1 CUT&RUN peak density map of sorted CD41+HSCs and CD41-HSCs for Runx1F/F and Runx1–/–genotypes. The map shows that there is no detectable binding of the anti-RUNX1 antibody to the genome of Runx1−/− genotype HSCs. (B) Motif enrichment analysis in HOMER database by the CisBP and de novo strategy. Different motifs were found enriched in CD41+HSC and CD41-HSC subpopulations. CD41+HSCs are enriched for Ets1, Fli1 and RUNX1, while CD41−HSCs are enriched for Sox4, Sox7 and Ets1. (C) RUNX1 CUT&RUN assay identifies the location of RUNX1 bounded peaks in sorted CD41+HSCs and CD41-HSCs. (D) Overlapping genes between RUNX1 bound targets identified by CUT&RUN peaks at upstream/promoter region and DEGs identified in CD41+HSCs and CD41−HSCs cells of Runx1–/–genotype through RNAseq. (E) Heatmap of the enriched RUNX1 target genes that are related to platelet activation signaling. (F) Representative RUNX1 Cut&Run tracks of Itga2b, Spi1, Selp and Tgfbr1 in Runx1F/F vs Runx1–/–CD41+HSCs generated by genome browser. Red boxes label the changed peaks in the gene bodies.

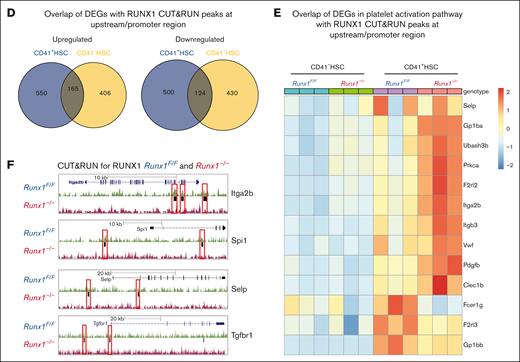
Next, we overlapped RUNX1 bound genes found in the CUT&RUN assay with DEGs of CD41+ and CD41– HSCs in the RNAseq data shown in Figure 5. In total, 1428 genes that were potential RUNX1 targets in CD41+ HSCs were identified; among the 715 upregulated DEGs in CD41+ HSCs, 165 overlapped with CD41– HSCs upregulated DEGs, and among the 624 downregulated DEGs in CD41+ HSCs, 124 overlapped with CD41– HSCs downregulated DEGs (Figure 6D). Similar to the observation in the GSEA analysis of DEGs in the RNAseq data, the platelet activation pathway was also enriched in the RUNX1 target set in the CUT&RUN data (Figure 6E). Indeed, we detected a binding of RUNX1 on the bodies of platelet signaling related genes, such as Selp and Itga2b (Figure 6F; supplemental Figure 8-C). Among these potential targets, 18 increased and 3 decreased in expression in Runx1– CD41+ HSCs. The upregulated genes included Pdgfb, VWF, Itgb3, Itga2b, Selp, and Trpc6, whereas the downregulated genes included Gna12 and Fcer1g (Figure 6E). In particular, we detected Tgfbr1 (TGF-β receptor1) as a RUNX1 target–without RUNX1; RNAseq data revealed that Tgfbr1 expression is compensatory increased, and several TGF-β signaling activation genes were upregulated (Stat3, Smad5, Mapk9, and so on) and repressor genes (Fst, Bmp4, Smad6, and so on) were downregulated in RUNX1 deficient MK-HSCs, suggesting that RUNX1 may regulate TGF-β signaling in MK-HSCs (supplemental Figure 8D).47,48 These results indicate that RUNX1 is essential for maintaining proper MK and platelet regulatory gene expressions in MK-HSCs.
Discussion
As a transcription factor or cofactor, RUNX1 is a key regulator of HSCs. HSCs are heterogenous, because even phenotypically well-defined HSC subpopulations may be functionally heterogeneous differing in life span, metabolism, cell cycle status, and differentiation potential.49-54 Recently, several groups have identified a subpopulation of HSCs that can predominantly differentiate into MKs and platelets (MK-biased) without going through the conventional HSC-MPP-MEP differentiation hierarchy.1,55 These HSCs are molecularly and functionally distinct from other HSC subpopulations and appear primed for MK or platelet-regulatory gene expression with enhanced propensity for short- and long-term reconstitution of platelets.4 Phenotypically, this subset of HSCs is characterized by the expression of vWF and CD41, accounting for ∼60% total HSCs and responsible for up to 50% to 60% of MK progenies.3 To date, however, the molecular regulation of this subset of HSCs and its relation to disease remains largely unknown.
In this study, we hypothesize that RUNX1 is a regulator of MK-HSCs and has a role in their differentiation potential into MKs. In previous studies, the classical surface markers, often used to identify LT-HSCs, were observed to be dysregulated because of RUNX1 deficiency, and the surface markers combining CD34− Flt3− LSK were considered better for approximated functional activity of LT-HSCs.33 Here, we applied a more stringent gating strategy and used CD150+ in addition to CD34–Flt3− LSK to sort the LT-HSCs. Our investigation revealed that RUNX1 is required for direct differentiation of MK-HSCs to MK cells, and loss of RUNX1 leads to an increase in the CD41+ MK-HSC subcomposition. Further examination of the function of MK-HSCs via CFU, liquid culture, collagen-based culture, and single cell division assays showed that the MK-HSC differentiation to MK cells can occur within 1 cell division cycle, and RUNX1 deficiency completely blocks such direct differentiation by MK-HSCs. Considering MK-HSCs may support up to 50% the platelet production as reported,3 our work suggests that RUNX1 has an important role in platelet production from MK-HSCs via direct differentiation, contributing to thrombocytopenia observed upon RUNX1 mutation. Mouse model studies of RUNX1 deficiency in HSCs had been described previously, and earlier studies have revealed that all subpopulations of RUNX1 deficient LSK cells displaya G1 cell cycle delay and decrease apoptosis.33 RUNX1-deficient hematopoietic stem and progenitor cells have a slowed growth, low biosynthetic, small cell phenotype, and markedly reduced ribosome biogenesis.13 Although RUNX1 is known to be required for HSC differentiation toward MK, given the limitations of the available mouse model how MK-HSC to MK to platelet production is at work remains unclear.
The single cell division and paired daughter cell assay is useful in revealing that MK-HSCs can give rise to daughter MK-lineage cells within 1 cell division. RUNX1 deficiency diminishes the ability of MK-HSC to differentiate to MK cells in 1 cell division. scRNAseq confirms our morphological observation that the daughter cells with a MK-like large cell shape are of the MK lineage, whereas the small non-MK–like daughter cells are of stem or MPP lineages. Although gene expression in MK-HSCs has been described previously,3,40 no molecular determinants have been implicated in the MK-HSC regulation. Our findings that RUNX1 binds to the TSS, gene body, and upstream regions of key factors of the platelet and MK formation pathways and is required for MK-HSC to MK differentiation, position RUNX1 in a key regulatory network of MK-HSCs.
Identifying bonafide RUNX1 targets has been challenging, in part because of the limitations imposed by a lack of suitable RUNX1 specific antibodies and to the sometimes confusing role of RUNX1 in either activating or suppressing putative targets by direct binding to DNA or as a cofactor of a regulatory complex.56 We performed CUT&RUN assays in HSCs, especially MK-HSCs, using Runx1–/– cells as baseline controls. The results showed that several MK differentiation related genes (eg, Selp and Itga2b) are likely RUNX1 binding targets. A few previously reported RUNX1 targets such as Spi, Myb, and Hmga2 were also observed, whereas a few other previously reported RUNX1 targets, such as Mpo and Cdk1 were not detected, suggesting that RUNX1 target activity is cell type specific as previous data acquired from induced pluripotent stem cells or leukemia cell lines may not be involved in the HSC compartment.15,17,23,57 It is possible that RUNX1 regulates CD41+ HSCs and CD41– HSCs differently, and the CUT&RUN condition allows the detection of the common RUNX1 binding motifs, such as runx1, Pu.1, ets1, fli1 on genes in CD41+ HSCs while detecting only indirect RUNX1 binding motifs, such as sox4 and sox7 on genes in CD41− HSCs. Together with the GSEA analyses, which revealed different regulatory pathways in MK-HSCs and non-MK–HSCs, this observation certainly may reflect the differential role of RUNX1 in the 2 populations of HSCs. Further exploration of the cell-intrinsic signaling regulation in MK-HSCs directed by RUNX1 binding suggests that the Spi1gene encoding PU.1 is a direct target of RUNX1. Although PU.1 is known for regulating common myeloid progenitor differentiation to erythroid and MK lineages,58 it may also be involved in regulating the commitment from MK-HSCs to MKs by RUNX1. Interestingly, Tgfbr1 (encoding TGF-β receptor1) appears to be a RUNX1 binding target in MK-HSCs, suggesting that RUNX1 regulates TGF-β signaling in MK-HSCs which may affect the HSC microenvironment in the BM and has a role in the pathophysiology of FPD/AML disease.47,59 Given the multiple regulatory mechanisms RUNX1 engages through multiple molecular complexes as a direct transcription factor or a costimulator or cosuppressor and the large panel of the regulatory gene activities or binding activities detected in the MK-primed HSC differentiation, it is our interpretation that RUNX1 regulates the MK-HSC differentiation process through a set of genes rather than 1 gene or several genes. A fine tune of this set of MK or platelet regulatory genes by RUNX1 is important for maintaining the differentiation potential of MK-HSCs to MKs.
Germ line mutation in RUNX1 is closely linked to familial platelet disorder with associated myeloid malignancies, FPD/AML.9,60,61 Our study is the first to implicate RUNX1 in the MK-HSC differentiation ability to directly develop into MK cells, whereas without RUNX1 the non-MK–HSCs can still maintain the differentiation ability to develop to myeloid cells, indicating a specific role of RUNX1 in the MK-HSC cell development. Although RUNX1 has been implicated in previous studies in the HSC to MPP to MEP differentiation hierarchy and MK maturation, therefore as a driver of the FPD phenotype, our work raises a possibility that RUNX1 mutations may lead to thrombocytopenia via the dysregulation of MK-HSCs in addition to the contribution in the defects during MPP-MEP mediated MK maturation or differentiation.21 This intriguing possibility will need future examination in animal models and human patients with FPD bearing RUNX1 mutations, which may provide insights into developing more effective therapeutic strategies targeting RUNX1 mutations and related changes not only in FPD but also in patients with somatic RUNX1 mutations.62-66 Whether some of the identified RUNX1 targets (eg, TGFB1) could be useful for FPD therapy will also be of interests for future studies.
Acknowledgments
The authors thank James Johnson for his technical assistance. The authors also thank the Research Flow Cytometry Core, Comprehensive Mouse and Cancer Core, DNA Core, and the Pathology Core at Cincinnati Children's Hospital Medical Center for their services and technical support.
This work was partially supported by grants U54 DK126108 from the National Institute of Diabetes, Digestion and Kidney Diseases, R01 CA234038 and R01 CA204895 from the National Cancer Institute, and R01 HL147536 from the National Institute of Heart, Lung and Blood of the National Institutes of Health, and a CancerFree Kids award.
Authorship
Contribution: C.W. and Y.Z. designed the study; C.W. performed experiments, interpreted data, and generated the figures; Z.T., X.C., W.W., K.N., A.P., J.W., F.L.H., A.K.D., and P.D. performed experiments; G.H. and H.G. interpreted data; C.W. and Y.Z. wrote the manuscript; and Y.Z. provided funding support.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yi Zheng, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Ave, Cincinnati, OH 45229; e-mail: yi.zheng@cchmc.org.

