

Key Points
Soft fibronectin–coated substrates favor MK adhesion and proplatelet formation through interaction with β3 but not with β1 integrins.
Stiff substrates promote fibronectin fibrillogenesis and increase intracellular contractility that prevents proplatelet formation.

Abstract
Megakaryocytes (MKs) are the precursor cells of platelets, located in the bone marrow (BM). Once mature, they extend elongated projections named proplatelets through sinusoid vessels, emerging from the marrow stroma into the circulating blood. Not all signals from the microenvironment that regulate proplatelet formation are understood, particularly those from the BM biomechanics. We sought to investigate how MKs perceive and adapt to modifications of the stiffness of their environment. Although the BM is one of the softest tissue of the body, its rigidification results from excess fibronectin (FN), and other matrix protein deposition occur upon myelofibrosis. Here, we have shown that mouse MKs are able to detect the stiffness of a FN-coated substrate and adapt their morphology accordingly. Using a polydimethylsiloxane substrate with stiffness varying from physiological to pathological marrow, we found that a stiff matrix favors spreading, intracellular contractility, and FN fibrils assembly at the expense of proplatelet formation. Itgb3, but not Itgb1, is required for stiffness sensing, whereas both integrins are involved in fibrils assembly. In contrast, soft substrates promote proplatelet formation in an Itgb3-dependent manner, consistent with the ex vivo decrease in proplatelet formation and the in vivo decrease in platelet number in Itgb3-deficient mice. Our findings demonstrate the importance of environmental stiffness for MK functions with potential pathophysiological implications during pathologies that deregulate FN deposition and modulate stiffness in the marrow.
Introduction
The daily production of 1011 blood platelets required to maintain normal hemostasis is made possible through unique mechanisms involving megakaryocytes (MKs). MKs are specialized giant cells derived from the differentiation of hematopoietic stem cells in the bone marrow (BM). Mature MKs interact with marrow sinusoid vessels and extend long cytoplasmic processes through the endothelial barrier.1 Proplatelets detach from the cell body to be further remodeled in the downstream microcirculation to release mature platelets.1,2
Although MKs grow well in suspension in vitro, they, nevertheless, reach a less extensive maturation state with poorly efficient proplatelet formation.3 Cells receive various signals from the environment ranging from soluble factors, cell-cell or cell-extracellular matrix (ECM) interactions and mechanical cues in vivo. It is known that adhesion to several ECM proteins, such as fibrinogen or fibronectin (FN), promotes proplatelet formation, whereas adhesion onto collagen I inhibits the process. However, these characteristics were observed upon the adhesion of MKs onto ECM-coated glass coverslip whose stiffness is physiologically irrelevant, and the proper impact of the substrate stiffness has been poorly investigated so far. The BM is composed mostly of hematopoietic cells and a few stromal cells, all tightly packed and surrounded by a loose meshwork of ECM,4 embedded in the trabeculae of the cancellous bone. The hematopoietic marrow is one of the softest viscoelastic tissue (E estimated < 0.5-24.7 kPa),5-7 being stiffer near the bone surface and softer near sinusoid vessels, which is the exclusive site of hematopoietic cell egress.8 These mechanical properties can change drastically during some pathological conditions. Chemotherapeutically induced myeloablation leads to a drastic, albeit transient, modification of the balance between cells and matrix proteins, whereas pathological conditions leading to marrow fibrosis increase tissue rigidity,9 emphasizing the need to understand how MKs react to various stiffness conditions.
It is now well -known that most cells adapt to tissue stiffness with responses that are cell-type dependent.10-13 Cells sense and react to different degrees of matrix stiffness through mechanotransduction mechanisms. Only a few studies suggested that MKs are sensitive to mechanical constraints, being able to respond to 3D confinement3 and collagen matrix stiffness.14,15 However, how MKs sense and react to FN-coated substrate stiffness in the same range as that of the BM is unclear. FN is an important matrix protein that polymerizes in a cell-dependent process to give rise to an insoluble, biologically active fibrillar conformation. FN matrix favors the deposition of other ECM proteins, such as collagens I, III, and IV and various other glycoproteins and thus plays a critical role in development of tissue fibrosis.16-21 FN from plasmatic origin is normally present throughout the marrow stroma and in the sinusoid vessel basement membrane.22,23 Importantly, MKs themselves also produce FN, which contribute to the creation of their proper niche24, and an increase in cellular FN protein deposition in the marrow stroma upon myelofibrosis has been reported.22 Thus, understanding the effects by which FN matrix stiffness influences MK behavior and functions is particularly clinically relevant. In this study, using mice as a model, we investigated the extent to which MKs were able to perceive substrate rigidity and notably explored the role of integrins as mechanosensors probing extracellular stiffness.25 Integrins, by exerting traction forces on the surrounding ECM, adjust adhesion strength via the modulation of adhesion structure composition and remodeling of the cytoskeleton. Each integrin subtype produces specific signals, leading to a unique response to the mechanics of its surroundings. Here, we found that although a stiff matrix promoted MK spreading and FN fibrillogenesis, a soft matrix that maintained a low intracellular contractility was preferred for adhesion and proplatelet formation. We also showed that FN stiffness-sensing and soft matrix–promoted proplatelet formation depends on β3 rather than β1 integrins.
Material and methods
Materials and detailed methods are presented in supplemental Material and Methods.
Mice
Animal experiments were conducted in accordance with European law, recommendations of the ethical committee, and national agreement. All mice had a C57BL/6 background and were aged between 3 and 4 months. The β1fl/fl26 and β3fl/fl27 mice were crossed with mice expressing the Cre recombinase under the control of the Pf4 promoter28 to obtain inactivation in the MK lineage.
MKs culture, adhesion, and proplatelet formation
Culture of lineage-negative mouse progenitors from femurs and tibia was performed as described.29 Polydimethylsiloxane (PDMS) substrates were prepared by mixing silicone base and curing agent in various ratios. The viscoelastic properties were characterized using rheology (supplemental Methods; supplemental Figure 1). For quantification of cell adhesion and proplatelet formation, we used FN circular patterns on the various PDMS substrates, coated with FN 50 μg/mL. For FN remodeling observation, we used biotin-labeled FN (50 μg/mL). MKs (5000 cells) were deposited on the micropatterns (50 μL) and counted 2-hours after seeding after a gentle wash, directly by the observation under the microscope of living cells. Evaluation of MK spreading over 5 hours was measured either using images obtained from living cells via bright field microscopy (Motic Image Plus 2.0 software) (apparent MK surface) or after fixation and immunolabeling (phalloidin-labeled spreading area). The proportion of MK extending proplatelets on PDMS was directly counted on living cells under the microscope 24 hours after MK seeding. As proplatelets frequently detached, MKs presenting only 1 proplatelet extension was considered as MK forming proplatelets, whereas free floating proplatelets were not taken into account. The proportion of MK extending proplatelets in liquid culture was counted on day 4 of the lineage-negative culture as described.29 The proportion of proplatelet formation in the explant culture was performed as described.30
Immunostaining, confocal image acquisition, and quantification
Five hours after seeding, MKs were washed and fixed using 4% paraformaldehyde. Immunolabeling was performed with primary antibodies followed by secondary antibodies and/or Alexa Fluor–labeled phalloidin and 4′,6-diamidino-2-phenylindole. Images were acquired using a Leica SP8 inverted confocal microscope. MKs were selected randomly, based solely on the large and polylobed 4′,6-diamidino-2-phenylindole (DAPI)–labeled nucleus, characterizing mature MKs, without prior observation of the other labeling, to be analyzed. To discriminate FN substrate remodeling from endogenously synthesized and released FN, biotinylated FN was coated on the PDMS substrates and revealed using fluorescein isothiocyanate–labeled streptavidin.
Electron microscopy
Electron microscopy was performed on intact flushed BM as previously described3 and is detailed in supplemental Materials and Methods.
Flow cytometry
Platelet integrins labeling was performed on whole blood anticoagulated with 6 mM EDTA via flow cytometry (Fortessa, Becton Dickinson). The mean fluorescence intensity was determined for each integrin subtype.
Statistics
Data are presented as mean ± standard error of the mean. Statistical analyses were performed using GraphPad Prism software version 9.2.0. Comparison among ≥3 groups were performed using ordinary 1-way analysis of variance (ANOVA) with Tukey multiple comparison tests. In some experiments, 2-way ANOVA analysis and Šidák multiple comparisons test were performed. Comparison between 2 groups were performed using unpaired 2-tailed t test. ∗P < .05; ∗∗P < .01; ∗∗∗P < .0001; ∗∗∗∗P < .00001; nonsignificant P > .05.
Results
MKs are able to sense and adapt to FN matrix stiffness
In order to evaluate the stiffness-sensing behavior of MKs, mature MKs were seeded onto PDMS substrates of various stiffnesses coated with FN. Here, we used a stiffness range (Young modulus ranging from 1.5 to 90 kPa) that encompasses normally soft and pathologically stiffer fibrotic BM tissues and compared these with glass coverslips that are commonly used for MK adhesion studies (Young modulus ∼70 Gpa; see “Materials and Methods”; supplemental Figure 1). The adhesion capacity was evaluated using FN-coated micropatterned surface (diameter 200 μm) 2-hours after MK seeding, with MKs still not being fully spread at that time. We observed a gradual decrease in the number of adhered MKs when stiffness increased, suggesting that MKs adhesion is promoted with a soft matrix (Figure 1A). In the presence of blebbistatin, MKs behaved similarly irrespective of the matrix stiffness and to the same extent as adhesion onto soft matrix (Figure 1B). This confirms that preferential adhesion onto soft matrix results from a stiffness-sensing mechanism and indicates that a low intracellular contractility favors their adhesion. For the spreading capacity evaluation, MKs were allowed to adhere for 5 hours. We observed that a higher stiffness led to more spreading (Figure 1C). As observed via the bright field microscopy of living cells, the surface of MK cells demonstrated a threefold increase when cultured on glass, compared with that when grown on a soft matrix (Figure 1C,D).

MKs sense FN-coated substrate stiffness. (A) MK adhesion for 2 hours onto PDMS substrate of various stiffness coated with FN 50 μg/mL using microcontact printing (circular patterns of 200 μm diameter). (Left) bright field image illustrating MK adhesion, bar is 50 μm; (right) quantification of the number of MK adhered per unit surface. (B) Effect of blebbistatin (100 μM) on MK adhesion. (A-B) Mean ± standard error of mean (SEM). Dots represent independent experiments (mean of 6-9 replicates); 2-way ANOVA analysis and Šidák multiple comparisons test, § indicates P < .0001, comparing the control condition with same substrate stiffness. (C-D) MK adhered for 5 hours and observed by bright field microscopy; (C) measurement of the visible surface of each MK measured; mean ± SEM, n = 3 independent experiments; statistics, ordinary 1-way ANOVA with Tukey multiple comparisons test. (D) Photomicrographs illustrating bright field microscopy observations; representative of at least 8 independent experiments; scale bar, 20 μm.
MKs sense FN-coated substrate stiffness. (A) MK adhesion for 2 hours onto PDMS substrate of various stiffness coated with FN 50 μg/mL using microcontact printing (circular patterns of 200 μm diameter). (Left) bright field image illustrating MK adhesion, bar is 50 μm; (right) quantification of the number of MK adhered per unit surface. (B) Effect of blebbistatin (100 μM) on MK adhesion. (A-B) Mean ± standard error of mean (SEM). Dots represent independent experiments (mean of 6-9 replicates); 2-way ANOVA analysis and Šidák multiple comparisons test, § indicates P < .0001, comparing the control condition with same substrate stiffness. (C-D) MK adhered for 5 hours and observed by bright field microscopy; (C) measurement of the visible surface of each MK measured; mean ± SEM, n = 3 independent experiments; statistics, ordinary 1-way ANOVA with Tukey multiple comparisons test. (D) Photomicrographs illustrating bright field microscopy observations; representative of at least 8 independent experiments; scale bar, 20 μm.
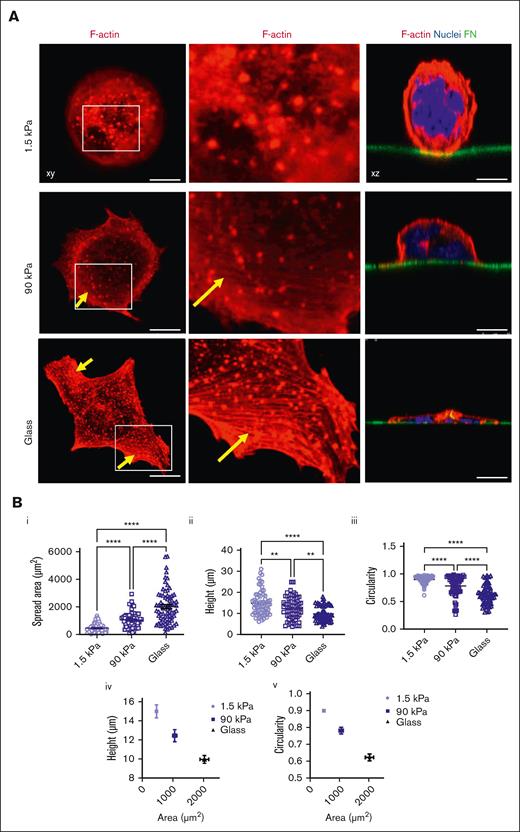
Matrix stiffness modulates the morphology of adhered MK and their F-actin organization
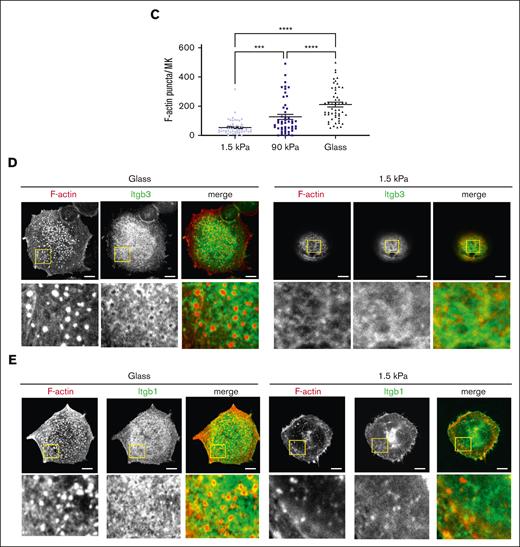
At the intracellular level, we observed that stiffer matrixes increased F-actin–positive spread area and cell flattening while decreasing circularity, with more spreading being correlated with lower height and lower circularity (Figure 2A,B; supplemental Figure 2). F-actin cytoskeleton organization at the basal side was mostly organized into F-actin puncta, whose number per cell increased with the increase in stiffness. F-actin fibers, which were always thin rather than in the form of large cables, were quasi-absent from soft substrates and became more numerous when adhered onto stiffer matrixes (Figure 2A,D,E). It has been described that MKs form podosomes when adhering onto matrix-coated glass.31 Similarly, we observed here that F-actin was mostly organized as puncta, whose number increased on stiff substrates. On glass and stiff substrates, podosome-like structures were characterized by central F-actin puncta surrounded by rings composed of β3 or β1 integrins, the 2 major β integrin subtypes expressed by MK (Figure 2D,E, left).32-35 Notably, not all MKs presented this typical integrin organization. Contrastingly, soft matrix did not promote this organization, with integrin being either independent of F-actin puncta or colocalized with mostly fuzzy F-actin (Figure 2D,E, right). Overall, this indicates that the organization/composition of F-actin adhesions varies based on the stiffness of the substrate.
Stiff matrix promotes higher MK spreading and F-actin organization. (A) MK spread at 5 hours on different substrate stiffness; (left), F-actin labeled in red; (middle), close-up view of the left images; (right) the nucleus is labeled in blue and FN-coated surface is labeled in green (in xz sections). Note the presence of F-actin puncta for all the stiffness with some F-actin fibers in stiff and glass substrate (arrows). Representative of at least 8 experiments. (B) Quantification of (i) F-actin positive spread area per each MK (n = 66-73 MKs), (ii) cell height (n = 66-73) and (iii) circularity (n = 66-73 MKs) (all from 3 independent experiments); individual values with mean ± SEM; (iv) and (v) co-relation between height (iv) or circularity (v) and F-actin positive area, mean ± SEM. (C) Quantification of F-actin puncta per MKs; individual values with mean ± SEM, n = 3 independent experiments. (D) Itgb3 (green) and F-actin labeling (red) in MKs adhered on glass (left) and on soft 1.5 kPa surface (right). E) Itgb1 (green) and F-actin labeling (red) in MKs adhered on glass (left) and 1.5 kPa soft surface (right). Observations in at least 3 independent experiments. (B-C) Ordinary 1-way ANOVA with Tukey multiple comparisons test. Scale bars, 10 μm (A,D,E).
F-actin adhesions are organized in response to integrin outside-in signaling after engagement. Accordingly, increased autophosphorylation of focal adhesion kinase (FAK) (pY397-FAK) at F-actin puncta, and a higher level of RhoA activation was observed as the stiffness increased (Figure 3A,B), suggesting enhanced signaling on stiffer matrixes. Addition of Mn2+ to ensure that integrins were in their activated ligand-binding state was not sufficient to promote complete spreading on soft substrates, suggesting that the difference in spreading between soft and stiff relies on the differences in stiffness sensing rather than on integrin-FN binding affinity (supplemental Figure 3).

Stiff matrix promotes higher intracellular signaling. (A) (Left) Higher FAK phosphorylation upon MK adhesion on stiff substrates. Red, F-actin; green, P-FAK. Scale bars, 10 μm. (Right) Quantification of P-FAK positive puncta per MK; mean ± SEM (20-25 MKs, 2 independent experiments). (B) (Left), RhoA–guanosine triphosphate immunolabeling. Scale bars, 15 μm. (Right) Higher RhoA activity upon MK adhesion on stiff substrate; mean ± SEM (53 MKs for 1.5 kPa, 69 MKs for 90 kPa, and 27 MKs for glass surface; 3 independent experiments). Ordinary 1-way ANOVA with Tukey multiple comparisons test.
Stiff matrix promotes higher intracellular signaling. (A) (Left) Higher FAK phosphorylation upon MK adhesion on stiff substrates. Red, F-actin; green, P-FAK. Scale bars, 10 μm. (Right) Quantification of P-FAK positive puncta per MK; mean ± SEM (20-25 MKs, 2 independent experiments). (B) (Left), RhoA–guanosine triphosphate immunolabeling. Scale bars, 15 μm. (Right) Higher RhoA activity upon MK adhesion on stiff substrate; mean ± SEM (53 MKs for 1.5 kPa, 69 MKs for 90 kPa, and 27 MKs for glass surface; 3 independent experiments). Ordinary 1-way ANOVA with Tukey multiple comparisons test.
Matrix stiffness directs the extent of MK-mediated FN reorganization
The presence of podosome-like structures suggest an increased possibility of MKs being prone to remodel the underlying FN matrix. Indeed, local FN accumulation was observed at the periphery of MKs (Figure 4). This FN accumulation resulted from the remodeling of the coated biotin-labeled FN. FN was assembled either at anchorage points, accumulated around the periphery of the cell, or formed radial arrays perpendicular to the cell edge. These radial arrays sometimes, but not always, colocalized with F-actin and integrins (Figure 4A-C). FN reorganization was different based on the stiffness of the matrix. On softer surfaces, FN was mostly forming aggregates. Increasing stiffness led to the appearance of longer and more individualized FN fibrils that reached a maximal length of ∼4 or 5 μm (Figure 4A-C). FN internalization does not seem to be a major event for fibrillogenesis because only a little amount of biotinylated FN was observed inside the MKs, although it was more on stiff matrix than on softer ones, in agreement with the longer fibrils observed on stiff substrates (Figure 4D).

Adhered MKs remodel matrix-bound FN. (A) confocal images showing streptavidin-FITC labeling of MKs having accumulated biotinylated FN at their periphery onto soft matrix and having remodeled into distinct fibrils onto stiff matrix at 5 hours; biotin-FN (green), F-actin (red); representative of at least 6 experiments; Scale bar, 10 μm. (B) Quantification of the proportion of MKs associated with peripheral FN assembly or distinct fibrils. Mean ± SEM, data are from 6 or 7 independent experiments. (C) Confocal images showing the localization of FN remodeling with that of Itgb3 (left) or Itgb1 (right); representative of at least 3 independent experiments. Scale bar, 10 μm. (D) FN internalization shown by streptavidin labeling of the substrate-bound biotinylated FN. Left, xz confocal images of MKs after 2 and 5 hours adhesion onto 1.5 and 90 kPa; (right) quantification of total fluorescence present inside MKs (arbitrary units) performed on maximal projections of image stacks above the substrate.
Adhered MKs remodel matrix-bound FN. (A) confocal images showing streptavidin-FITC labeling of MKs having accumulated biotinylated FN at their periphery onto soft matrix and having remodeled into distinct fibrils onto stiff matrix at 5 hours; biotin-FN (green), F-actin (red); representative of at least 6 experiments; Scale bar, 10 μm. (B) Quantification of the proportion of MKs associated with peripheral FN assembly or distinct fibrils. Mean ± SEM, data are from 6 or 7 independent experiments. (C) Confocal images showing the localization of FN remodeling with that of Itgb3 (left) or Itgb1 (right); representative of at least 3 independent experiments. Scale bar, 10 μm. (D) FN internalization shown by streptavidin labeling of the substrate-bound biotinylated FN. Left, xz confocal images of MKs after 2 and 5 hours adhesion onto 1.5 and 90 kPa; (right) quantification of total fluorescence present inside MKs (arbitrary units) performed on maximal projections of image stacks above the substrate.
β3 integrin is the main integrin for FN stiffness sensing, whereas both β1 and β3 contribute to fibrillogenesis
We then questioned which integrin receptors were involved in MK stiffness sensing and FN remodeling. MKs express several integrins acting as FN receptors.36 In the BM, FN was regularly found surrounding MKs and in contact with β1 and β3 integrin subunits (Figure 5A). Upon its adhesion to FN, we found 3 times more Itgb3 than Itgb1 present on the basal side, in contact with the matrix. There, Itgb3 was mostly colocalized with the Itgb1 subunit (supplemental Figure 4). To evaluate their respective roles, we resorted to use MKs from β1- and β3-deficient mice (supplemental Figure 5). Absence of β1 integrin had no effect (Figure 5B,C). In striking contrast, the absence of β3 integrin strongly modified the capacity of MKs to react to stiffness. The number of adhering Itgb3–/– MKs on soft surface was leveled down to that on stiff surface, indicating that soft matrix–promoted adhesion depends on β3 integrin rather than on β1 (Figure 5B). Further, the increased spreading promoted by the stiff substrate was impaired in β3 integrin–deficient MKs (Figure 5C). No additional effect was observed with a double inactivation of these 2 integrin subunits. At the intracellular level, Itgb3–/–prevented glass coverslip-mediated F-actin fibers formation, increased the spreading area and decreased circularity as well as RhoA activation and FAK phosphorylation, whereas Itgb1 inactivation had no impact (Figure 5D,E). Overall, these data indicate that FN-coated substrate stiffness sensing depends mainly on β3 integrin. Furthermore, we questioned which of the αvβ3 and αIIbβ3 integrins were involved in the process. We measured the MK spread area in the presence of either a blocking anti-αv antibody or integrilin that blocks αIIbβ3. As shown in supplemental Figure 6, αIIbβ3 was the integrin responsible for stiffness sensing because blocking αvβ3 had no impact, whereas integrilin decreased both the spreading on stiff substrate and proplatelet formation on soft substrate in control MKs, to a value similar to that of Itgb3–/– MKs.

Integrin β3 is required for FN stiffness sensing and fibrillogenesis. (A) Immunolabeling of mouse BM sections showing co-localization between FN (green) and Itgb3 (left) or Itgb1-positive MK (right) (red labeling). Representative of at least 3 independent sections. Bar is 10 μm. (B) Number of WT, Itgb1–/–, Itgb3–/–, and Itgb1b3–/– MK adhered for 2 hours onto PDMS substrates of various stiffness coated with FN 50 μg/mL using microcontact printing (circular patterns of 200 μm diameter); 3 independent experiments performed simultaneously with the 4 genotypes, each dot represents the mean of 5 replicates; ordinary 1-way ANOVA with Tukey multiple comparisons test. (C) Measurement of the visible surface of MK adhered for 5 hours, observed by bright field microscopy. Ordinary 1-way ANOVA with Tukey multiple comparisons test; §P < .001 comparing the control with the substrate of same stiffness. (D) (i) F-actin labeling of WT and Itgb3–/– MK adhered on FN-coated glass coverslip for 5 hours. Scale bar, 10 μm; (ii) positive F-actin spreading area per MK; (iii) circularity; (E) (Left) Confocal images showing immunofluorescence labeling (arbitrary units) of RhoA-GTP (yellow) and P-FAK (green) in control, Itgb1–/– and Itgb3–/– MKs following 5 hours adhesion onto soft and stiff FN substrate; (middle) quantification in arbitrary units of the RhoA-GTP labeling intensity; n = 30 to 46 MKs from 3 independent experiments; (right) quantification of the number of P-FAK positive puncta per MK; n = 28 to 31 MKs from 3 independent experiments. Ordinary 1-way ANOVA with Šidák multiple comparisons test. (F) (Left) Representative 2 confocal images showing biotin-FN (revealed by FITC-streptavidin, green) at the periphery of the control, Itgb1–/–, Itgb3–/–, and Itgb1b3–/– MKs adhered for 5 hours on 90 kPa substrate. Scale bar, 10 μm. (Right) Quantification of the proportion of MK presenting FN accumulation at anchorage points, peripheral accumulation and visible individual fibrils (n = 3 independent experiments; 60-63 MKs per genotype).
Integrin β3 is required for FN stiffness sensing and fibrillogenesis. (A) Immunolabeling of mouse BM sections showing co-localization between FN (green) and Itgb3 (left) or Itgb1-positive MK (right) (red labeling). Representative of at least 3 independent sections. Bar is 10 μm. (B) Number of WT, Itgb1–/–, Itgb3–/–, and Itgb1b3–/– MK adhered for 2 hours onto PDMS substrates of various stiffness coated with FN 50 μg/mL using microcontact printing (circular patterns of 200 μm diameter); 3 independent experiments performed simultaneously with the 4 genotypes, each dot represents the mean of 5 replicates; ordinary 1-way ANOVA with Tukey multiple comparisons test. (C) Measurement of the visible surface of MK adhered for 5 hours, observed by bright field microscopy. Ordinary 1-way ANOVA with Tukey multiple comparisons test; §P < .001 comparing the control with the substrate of same stiffness. (D) (i) F-actin labeling of WT and Itgb3–/– MK adhered on FN-coated glass coverslip for 5 hours. Scale bar, 10 μm; (ii) positive F-actin spreading area per MK; (iii) circularity; (E) (Left) Confocal images showing immunofluorescence labeling (arbitrary units) of RhoA-GTP (yellow) and P-FAK (green) in control, Itgb1–/– and Itgb3–/– MKs following 5 hours adhesion onto soft and stiff FN substrate; (middle) quantification in arbitrary units of the RhoA-GTP labeling intensity; n = 30 to 46 MKs from 3 independent experiments; (right) quantification of the number of P-FAK positive puncta per MK; n = 28 to 31 MKs from 3 independent experiments. Ordinary 1-way ANOVA with Šidák multiple comparisons test. (F) (Left) Representative 2 confocal images showing biotin-FN (revealed by FITC-streptavidin, green) at the periphery of the control, Itgb1–/–, Itgb3–/–, and Itgb1b3–/– MKs adhered for 5 hours on 90 kPa substrate. Scale bar, 10 μm. (Right) Quantification of the proportion of MK presenting FN accumulation at anchorage points, peripheral accumulation and visible individual fibrils (n = 3 independent experiments; 60-63 MKs per genotype).
The main integrin reported to mediate FN fibrillogenesis is α5β1. Interestingly, we found, here, that not only the absence of β1 but also β3 integrin altered the extent of remodeling and the number of MKs presenting ECM accumulation and fibrils (Figure 5F). Furthermore, increasing integrin affinity using Mn2+ increased the fibrillogenesis capacity for both Itgb1–/– or Itgb3–/– MKs (data not shown). Together, this suggests a partial redundant role for both receptors in the process of FN assembly, and in the absence of one subunit, the other takes over. Accordingly, the absence of both integrin subtypes virtually suppressed all remodeling (Figure 5F).
Soft matrixes promote proplatelet formation
The fate of all mature MKs is to extend proplatelets, which were evaluated 24 hours after the seeding of MKs. We observed that a soft matrix was more favorable, with ∼40% MKs extending proplatelets on 1.5 kPa compared with 20% of those extending proplatelets on a 90 kPa substrate (Figure 6A), and only a little percentage of them on FN-coated glass (Figure 6A). Interestingly, we observed that MKs that were well spread on glass surfaces from 4 to 5 hours, tended to become more rounded at 24 hours (Figure 6B; supplemental Movie 1), similar to MKs that adhered for 5 hours on soft matrix (compare Figure 2Biv to Figure 6B). This could reflect a reorganization of the cytoskeleton toward less contractility and be a prerequisite to allow proplatelet extension. Accordingly, decreasing the myosin activity via the addition of blebbistatin resulted in loss of stress fibers (not shown) and increased the proplatelet formation on stiff matrixes. This suggests that a stiff substrate, notably by promoting a very strong intracellular contractility, is detrimental for proplatelet formation (Figure 6C). The capacity of MKs to extend proplatelets after blebbistatin treatment was similar irrespective of substrate stiffness and intermediate between that observed for soft and stiff matrixes, indicating that an intermediate range of intracellular actomyosin is beneficial for proplatelet extension.

Soft matrix is preferred for proplatelet formation. (A) (Left) Bright field image of MK forming proplatelets after 24 hours adhesion on the 1.5 kPa FN micropattern. Scale bar, 50 μm. (Right) Quantification of MK forming proplatelets depending on the matrix stiffness. Ordinary 1-way ANOVA with Tukey multiple comparisons test, mean ± SEM, n = 3 independent experiments, each dot is the mean of 9 replicates. (B) Decreased spreading after 24 hours adhesion. (i) xy and xz view of a MKs adhered for 5 or 24 hours; red, F-actin, blue, nucleus. Scale bar, 10 μm; (ii) time-lapse of a MK expressing Lifeact-GFP (green); acquisition was one every hour and started 6 hours after seeding; bar is 10 μm. (iii) Co-relation between spreading area and height of MKs adhered for 5 hours or 24 hours on FN-coated glass surface. Mean ± SEM from 25 MKs in 2 independent experiments. (C) Impact of blebbistatin on proplatelet formation on the different substrate stiffness after 24 hours adhesion; n = 3 independent experiments; each dot is the mean of 9 replicates ± SEM. For the vehicle group, statistical analysis was performed with ordinary 1-way ANOVA and Tukey multiple comparisons; ∗∗∗∗P < .0001. Comparison between vehicle and blebistatin was with 2-way ANOVA and Šidák multiple comparisons; §P < .05, §§P < .01, comparison between vehicle and blebbistatin treatment for a same stiffness.
Soft matrix is preferred for proplatelet formation. (A) (Left) Bright field image of MK forming proplatelets after 24 hours adhesion on the 1.5 kPa FN micropattern. Scale bar, 50 μm. (Right) Quantification of MK forming proplatelets depending on the matrix stiffness. Ordinary 1-way ANOVA with Tukey multiple comparisons test, mean ± SEM, n = 3 independent experiments, each dot is the mean of 9 replicates. (B) Decreased spreading after 24 hours adhesion. (i) xy and xz view of a MKs adhered for 5 or 24 hours; red, F-actin, blue, nucleus. Scale bar, 10 μm; (ii) time-lapse of a MK expressing Lifeact-GFP (green); acquisition was one every hour and started 6 hours after seeding; bar is 10 μm. (iii) Co-relation between spreading area and height of MKs adhered for 5 hours or 24 hours on FN-coated glass surface. Mean ± SEM from 25 MKs in 2 independent experiments. (C) Impact of blebbistatin on proplatelet formation on the different substrate stiffness after 24 hours adhesion; n = 3 independent experiments; each dot is the mean of 9 replicates ± SEM. For the vehicle group, statistical analysis was performed with ordinary 1-way ANOVA and Tukey multiple comparisons; ∗∗∗∗P < .0001. Comparison between vehicle and blebistatin was with 2-way ANOVA and Šidák multiple comparisons; §P < .05, §§P < .01, comparison between vehicle and blebbistatin treatment for a same stiffness.
Integrin β3 engagement is critical for proplatelet formation
Next, we evaluated the role of integrin in soft matrix–promoted proplatelet formation. We found that β1 deficiency did not affect proplatelet formation (Figure 7A). In contrast, β3 integrin controlled proplatelet formation depending on the matrix stiffness, which is in line with its role in stiffness sensing, whereas inactivation of both β1 and β3 integrins had no further additional impact. This suggested that although stiff matrix inhibited proplatelet extension independently on integrins, soft matrix promotion of proplatelet extension depends on β3 integrin.

FN-β3 integrin interaction promotes soft matrix–mediated proplatelet formation. (A) Quantification of MK forming proplatelets depending on the matrix stiffness and genotype; mean statistics: 1-way ANOVA and Tukey multiple comparisons, n = 3 independent experiments, each dot is the mean of at least 9 replicates ±SEM; §§P < .01; §§§P < .001; §§§§P < .0001 comparing the control with substrates of same stiffness. (B) Proplatelet formation by MK grown in liquid culture; (i) bright field observation of MK forming proplatelets; arrows indicate proplatelets; (ii) quantification of the proportion of Itgb3–/– MKs extending proplatelets vs control MKs; mean ± SEM, no significant difference (t test), n = 5 independent cultures; (iii) quantification of the proportion of Itgb1b3–/– MKs extending proplatelets vs control MKs; mean ± SEM, no significant difference; n = 3 independent cultures. (C) BM explant experiment. (Left) Phase contrast observation of MK forming proplatelets at the periphery of the tissue explant; arrows indicate proplatelets. (Right) Quantification of proplatelet formation. Mean ± SEM, statistics, 1-way ANOVA and Tukey multiple comparisons, n = 10 to 16 independent marrows. (D) Mouse platelet count. One-way ANOVA and Tukey multiple comparisons, mean ± SEM, n = 10 to 20 mice. (E) (i), BM sections of control and Itgb1b3–/– mice immunolabeled for MKs von Willebrand Factor vWF, red and for vessels (FABP4, green); representative of at least 3 marrow sections. (ii) quantification of MKs per mm2; mean ± SEM, n = 5 mice, 3 sections per marrow and 4 fields of observation per section; (iii), distance between MKs and sinusoid vessels; mean ± SEM; n = 5 mice; 3 sections per marrow and 3 to 4 fields of observation per marrow section. No significant difference (t test).
FN-β3 integrin interaction promotes soft matrix–mediated proplatelet formation. (A) Quantification of MK forming proplatelets depending on the matrix stiffness and genotype; mean statistics: 1-way ANOVA and Tukey multiple comparisons, n = 3 independent experiments, each dot is the mean of at least 9 replicates ±SEM; §§P < .01; §§§P < .001; §§§§P < .0001 comparing the control with substrates of same stiffness. (B) Proplatelet formation by MK grown in liquid culture; (i) bright field observation of MK forming proplatelets; arrows indicate proplatelets; (ii) quantification of the proportion of Itgb3–/– MKs extending proplatelets vs control MKs; mean ± SEM, no significant difference (t test), n = 5 independent cultures; (iii) quantification of the proportion of Itgb1b3–/– MKs extending proplatelets vs control MKs; mean ± SEM, no significant difference; n = 3 independent cultures. (C) BM explant experiment. (Left) Phase contrast observation of MK forming proplatelets at the periphery of the tissue explant; arrows indicate proplatelets. (Right) Quantification of proplatelet formation. Mean ± SEM, statistics, 1-way ANOVA and Tukey multiple comparisons, n = 10 to 16 independent marrows. (D) Mouse platelet count. One-way ANOVA and Tukey multiple comparisons, mean ± SEM, n = 10 to 20 mice. (E) (i), BM sections of control and Itgb1b3–/– mice immunolabeled for MKs von Willebrand Factor vWF, red and for vessels (FABP4, green); representative of at least 3 marrow sections. (ii) quantification of MKs per mm2; mean ± SEM, n = 5 mice, 3 sections per marrow and 4 fields of observation per section; (iii), distance between MKs and sinusoid vessels; mean ± SEM; n = 5 mice; 3 sections per marrow and 3 to 4 fields of observation per marrow section. No significant difference (t test).
To rule out a possible proplatelet formation defect that could result from β3 integrin deficiency independently on matrix interaction, we examined proplatelet formation in liquid culture. In striking contrast with the presented data, proplatelet formation in liquid culture was not impaired by Itgb3–/– deletion, showing that under these conditions, Itgb3 is dispensable for proplatelet formation (Figure 7B). We then evaluated proplatelet formation in an ex vivo model of BM explant culture, similar to native conditions in which MKs interact with the ECM.30 In these conditions, the proplatelet formation rate was decreased by half in MKs from Itgb3–/– or Itgb1b3–/–- explant marrow (Figure 7C), consistent with our data on soft FN-coated substrates (Figure 7A). Furthermore, it supports the importance of β3 integrin engagement in controlling proplatelet formation. Accordingly, in vivo circulating platelet count had decreased by 30% in Itgb3–/–- and 55% in Itgb1b3–/– mice, respectively (Figure 7D). This decreased platelet number is probably, largely, due to the defective proplatelet formation, because Itgb1b3–/– mice had normal amounts of hematopoietic progenitors, MK progenitors, and mature MKs (supplemental Figure 7; Figure 7E). In addition, we ruled out potential defects in MK migration toward sinusoid vessels (Figure 7E). The significantly lower platelet counts in Itgb1b3–/– compared with Itgb3 inactivation alone was surprising, considering the proplatelet data. In an attempt to understand this discrepancy between in vitro and in vivo conditions, we performed in situ observations using transmission electron microscopy. We sought to evaluate the close interaction between MKs and sinusoid vessels that are surrounded by a complex matrix and the level of MK maturation because we know that in vitro, MKs do not reach the same level of maturation as they do in vivo. Interestingly, we observed that although MK from Itgb1–/– and Itgb3–/– mice appeared normal at the ultrastructural level, inactivation of both integrins led to an abnormal organization of the demarcation membrane system (DMS), the reservoir for the elongation of proplatelets and the membrane of the future platelets (supplemental Figure 8). In these MKs, the DMS leaflets were abnormally stacked, sometimes forming circular stacks, or being unevenly distributed in the cytoplasm, suggesting that signaling through both integrins contributes to the normal DMS organization. This abnormal DMS stacking possibly affects the availability of the membrane reservoir and, ultimately, the total number of released platelets per MK.
Discussion
MK normally reside in a very soft environment whose stiffness can be modulated depending on pathological conditions, questioning the extent to which they respond and adapt to stiffness modifications. We found that MKs sense the stiffness of FN-coated substrates that influences their adhesion and spreading, the organization of F-actin cytoskeleton, and capability to remodel FN into fibrils and extend proplatelets. The marrow is a very soft tissue embedded in the stiff bone having a stiffness in the Gpa range. We, therefore, chose to evaluate the behavior of MKs using substrate stiffnesses ranging from 1.5 kPa to 90 kPa and glass coverslips to surround stiffness that can be perceived within the marrow. We observed that the stiffness threshold detected by MKs depended on the behavior examined, between 12 and 90 kPa for adhesion and >90 kPa for spreading and proplatelet formation. One unexpected and counterintuitive finding in the mechanobiology field was the adherence of MKs preferentially to softer matrixes, whereas most cells usually adhere better on stiffer matrixes. This may reflect the fact that their normal environment is naturally soft,6,7 and we also found that soft matrix was preferred over stiffer ones (10, 30, 90 kPa, and glass) for the extension of proplatelets. Accordingly, it has been shown notably with neuronal cells that matching the substrate stiffness in vitro with that of target tissue stiffness in vivo allows for a better approach toward understanding the characteristics and phenotypes of native cells.12,37,38 It is also possible that this property contributes to the normal hematopoietic development. In the embryo, MKs first develop in the embryonic liver, a very soft tissue39, and then as the fetal liver matures toward its definitive hepatic function, its stiffness increases,40 which may contribute to render the environment less hospitable for MK progenitors.
We showed that soft matrixes promoted lower MK intracellular contractility compared with stiff matrixes, as observed with the lower levels of RhoA activation and F-actin fibers formation, which likely results in less force across the ECM-integrin-cytoskeleton linkage. We may hypothesize that the higher intracellular contractility is detrimental for proplatelet extension. Accordingly, decreasing the intracellular contractility by adding blebbistatin when MKs were on stiff surface increased proplatelet formation. This is also in accordance with previous studies showing that the RhoA/Rhokinase/MyosinIIA axis is inhibitory for proplatelet formation.15,41-44 Hence, substrates in the same range of stiffness as that of the BM, by minimizing internal stress, are preferred for optimal proplatelet formation. Interestingly, we observed that on glass surface MKs that were well spread after 5 hours underwent rounding by 24 hours, suggesting the presence of some internal clock dedicated at decreasing intracellular contractility to allow proplatelet extension.
Upon adhesion to the FN, we discovered that MKs were able to remodel the substrate-bound FN matrix into basal fibrillar structures that surround the cells. Again, the extent of FN reorganization and morphology of the fibrils depended on the stiffness of the substrate, as also observed in other cells.45 A soft substrate led essentially to FN accumulation around the MKs. Upon adhesion on a stiffer matrix, a higher proportion of MKs exhibited longer and individualized FN fibrils, potentially leading to different functional properties. This difference is in line with the fact that FN assembly into fibrils requires application of cell-derived contractile forces to expose cryptic binding sites.19,46 FN fibrils represent the bioactive form of FN that induces mechanical and chemical signals and often precedes and promotes assembly of other matrix proteins.16,17,20 Therefore, alteration in BM environmental stiffness may create a feedback loop through which MK-mediated FN fibrillogenesis promotes collagen deposition and increases tissue stiffness, which, in turn, upregulates FN matrix synthesis or assembly. Reciprocally, FN fibrillogenesis has also been described after MK adhesion onto collagen I.24 Therefore, such bidirectional pathological feedback loops could contribute to the development of BM fibrosis, a pathological condition of neoplastic or nonneoplastic origin, which is characterized by an excess deposition of FN and collagens, associated with abnormal MK number and function and, ultimately, marrow failure.9,22,47
Integrins are known to differentially contribute to FN signaling and rigidity sensing through the cooperation between α5β1 and αvβ3.48 However, because each integrin type mediates distinct intracellular signaling resulting from different binding partners, this translates into differential cytoskeletal and force transmission.49-51 Therefore, depending on the integrin subunit equipment of a cell, adhesion structures may be functionally very dissimilar. Notably, it has been proposed that after its interaction with FN, α5β1 would be rather dedicated to force transmission, whereas αvβ3 would be the predominant mechanosensor.52-54 Here, we showed that MKs rely mostly on β3 subunit for stiffness sensing and proplatelet formation because β1 subunit inactivation does not affect adhesion, spreading, or proplatelet formation.
We can speculate that the reason why β3 is prominent over β1 could be the unique integrin expression profile of MKs. Quantifications in platelets previously showed that β3 subunit is 4- to 6-times more expressed than β1,34,35 and αIIbβ3 is estimated to be 80-times more represented than α5β1.35 Among the 2 β3 based integrins expressed by MKs, we showed that αIIbβ3 was the one responsible for stiff substrate–mediated spreading and soft matrix–mediated proplatelet formation. αvβ3 seems to be uninvolved here, perhaps, because of its low expression, reported to be less than 0.5% of that of αIIbβ3 on platelets.35 The manner in which β3 integrins of MKs act as mechanosensor upon adhesion to FN remains unclear and would be of interest for a more detailed future study. Especially, whether β3 integrin acts as a direct mechanosensor here, through protein binding to its tail that controls actomyosin contractility as shown in other cells,48,54,55 or another mechanoreceptor acts upstream to promote β3 integrin inside out is unclear. This indirect mechanism was shown for MK adhesion onto collagen IV, in which Trpv4 activates Itgb1,14 or onto erythroblasts, in which Piezo1 signals to activate α4β1 and α5β1.56
Interestingly, it was recently shown that expression of integrins α5, αIIb, and β3 subunits was elevated in MK in JAK2V617F+ mice, a model mimicking myelofibrosis, whereas β1 and α2 subunits remained unchanged.57 This suggests that during the development of myelofibrosis, the cards are completely reshuffled regarding the relative expression of integrins. One can imagine that this is an adaptation of MKs to a new, abnormally matrix-rich environment.
Considering FN assembly, we showed here that MKs rely on both β1 and β3 integrins, whereas the absence of both integrin subunits accordingly abrogates fibril formation. Fibrillogenesis mediated by fibroblasts or mesenchymal cells has been essentially ascribed to integrins bearing the β1 subunits, notably the typical α5β1 integrin.58 However, in a number of β1-null cells, αvβ3 can partly contribute to FN assembly.59 In Chinese hamster ovary cells expressing αvβ3 and αIIbβ3, stimulation of the integrins promotes FN matrix assembly.60,61 Recently platelets were found to favor FN fibrillogenesis through αIIbβ3,62 and a recent study identified ICAP-1 as a candidate directly controlling Itgb3-mediated mechanosensitive responses including fibrillogenesis in osteoclasts,55 leading to the assumption that the type of integrin mediating FN fibrillogenesis depends on the relative expression level of the integrin and their associated proteins. Our study also points to the fact that it is the interaction between FN and β3 integrin on the soft substrate, but not on stiffer ones, that promotes proplatelet formation. Indeed, no defect was observed in liquid culture in which integrin is presumably not engaged with the matrix protein. In the explant model, the glass coverslip is not coated with matrix protein but importantly, MKs have grown in their native matrix-rich environment, including FN, likely explaining why proplatelet formation is affected by the absence of Itgb3. Soft FN substrate initiates a weaker integrin-mediated intracellular signaling compared with stiffer matrixes, as analyzed with FAK phosphorylation, RhoA activation, and F-actin organization. We may, thus, hypothesize that this weak signaling is more beneficial for normal proplatelet extension by MKs than stronger signaling.
Finally, the role of β3 integrin in proplatelet formation can be in fine translated to the production of platelets. This occurs in mice in which the number of circulating platelets in Itgb3-deficient mice is decreased, as previously reported by others.63 In human, a number of mutations in integrin αIIbβ3 that lead to a decrease in its expression level, are responsible for a rare autosomal recessive bleeding disorder named Glanzmann thrombasthenia. Although having no overt thrombocytopenia, platelet count of patient with Glanzmann thrombasthenia is at the lower end of the normal range.64 More importantly, autosomal dominant αIIbβ3 mutations lead to macrothrombocytopenia. This phenotype has been assigned to impaired cytoskeletal remodeling because of the constitutive integrin signaling,65-68 in line with our observation of low proplatelet formation on stiff surface that promotes higher signaling and intracellular contractility.
The lower platelet number observed in the double knockout compared to Itgb3–/– alone was unexpected in view of the absence of additive effect on the proportion of MKs extending proplatelets in vitro or ex vivo. The observation of abnormal DMS organization in Itgb1b3–/– (supplemental Figure 7) could alter the normal process of membrane fueling upon proplatelet extension and this way contribute to decrease further the platelet yield. Whether this abnormal organization is directly linked to the absence of integrin, impairing matrix signaling toward DMS organization, or indirectly through disorganization of the cytoskeleton secondary to integrin deficiency, is still unclear.
In conclusion, we present evidence that MKs, though being anchorage-independent cells, are able to sense stiffness of a FN-coated substrate. We found, here, that although stiff matrixes promoted higher intracellular signaling and F-actin organization leading to increased spreading and FN fibril formation, soft substrates that mirror the physiological BM softness favor MK adhesion and proplatelet formation through interaction with β3 integrin, contributing in this manner to platelet biogenesis. This study was performed with mouse MKs, and similar studies should be conducted with human MK progenitors to confirm that these observations also apply to human cells. In that case, these data may be of particular relevance for pathologies associated with increased or decreased modifications in marrow tissue stiffness.
Acknowledgments
The authors warmly thank Martial Balland for all the fruitful discussions and sound advice. The authors also thank Patricia Laeuffer and Noémie Gros for excellent technical assistance, Fabien Pertuy who initiated the establishment of the mouse lines, and Monique Freund and all the animal facility of EFS Grand-Est for animal husbandry. I.G., L. Ruch, and this work were supported by Agence Nationale de la Recherche grant (ANR PlatForMechanics-18-CE14-0037). T.N. was the recipient of an IDEX fellowship from University of Strasbourg.
Authorship
Contribution: T.N. initiated the project; I.G. and T.N. designed and performed experiments and analyzed the data; N.B.-J., J.W., L. Ruch, L. Reininger, N.B. and D.C. performed and analyzed experiments; A.E. performed transmission electron microscopy; F.L. discussed results; and C.L. conceived the project and designed the research, analyzed data and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing interests.
Correspondence: Catherine Léon, UMR_S1255 INSERM-Université de Strasbourg-Etablissement Français du Sang (EFS), 10, Rue Spielmann, BP No. 36, 67065 Strasbourg Cedex, France; e-mail: catherine.leon@efs.sante.fr.

