Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome characterized by extreme immune activation, resulting in pathologic inflammation. The diagnosis includes a spectrum of inherited or acquired defects in cytotoxic lymphocyte function, often with uncontrolled infections. HLH may also arise as the result of persistent antigen stimulation due to autoimmune disease or malignancy. HLH is often described in binary terms as “primary,” indicating Mendelian inheritance of gene mutations resulting in cytotoxic lymphocyte dysfunction, or “secondary” indicating an acquired reactive disorder. Increasing evidence describes HLH as more complex phenomenon, resulting from specific immune challenges in patients with a susceptible genetic background. Early recognition of HLH and evaluation of potential causes is critically important, as survival generally requires urgent treatment with immune suppression and resolution of the activating antigen. However, the diagnosis of HLH is challenged by the myriad of pathways that lead to pathologic inflammation and the clinical overlap with other conditions. Further improvements in therapy will require prospective trials to define optimal strategies for each patient based on the individual paths that lead to pathologic inflammation.
Learning Objectives
To understand the rationale for the diagnosis of hemophagocytic lymphohistiocytosis (HLH)
To review the mechanisms that lead to pathologic inflammation in HLH
To discuss strategies for recognition and treatment of pathologic inflammation in critically ill patients
Evaluation and discussion of hemophagocytic lymphohistiocytosis (HLH) is an intellectual challenge due to the imprecise definitions of the syndrome and overlap with more common presentations of inflammatory conditions. The diagnostic uncertainty adds practical challenges to treatment of patients with HLH in whom there is an urgent need to initiate potentially toxic (and clinically counterintuitive) immune suppression in the setting of uncontrolled infection. To successfully treat a patient with HLH, the condition must first be recognized. Survival is estimated to be <10% in patients with HLH who do not receive immunochemotherapy.1
What is HLH?
”Hemophagocytic lymphohistiocytosis” is an imperfect name for this syndrome (or group of syndromes) as “hemophagocytosis” is often absent and “lymphohistiocytosis” lacks precision. In general, HLH is a syndrome of pathologic immune activation with clinical manifestations of extreme inflammation. HLH was first recognized as an inherited immune disorder in 1952 as “familial hemophagocytic reticulosis” in siblings who both presented in early infancy with fever, cytopenias, hepato-splenomegaly, and coagulopathy.2 Interestingly, a trial of adrenocorticotropic hormone was given to the second child, with some temporary clinical improvement. She survived 94 days after the first symptoms, whereas her brother survived only 21 days. Hemophagocytosis was noted in lymph node and spleen by autopsies of both children. These initial cases are classic for inherited HLH, where a previously healthy infant or toddler presents with nonspecific symptoms of inflammation, and then rapidly becomes critically ill with multisystem organ failure. Even with infants, this is an extremely challenging diagnosis to make given the clinical overlap with more common conditions. In a review of the experience at Texas Children's Hospital, the majority of children ultimately diagnosed with gene-proven HLH were initially diagnosed with Kawasaki Disease (C.E.A. and K.L.M., unpublished observation). The diagnostic challenge is magnified in older children and adults in whom suspicion is much lower and range of alternative diagnoses is much broader. In general, HLH is differentiated from other intrinsic and reactive immune disorders by the degree of pathologic inflammation.
Diagnostic criteria
Diagnostic criteria were developed through consensus by the Histiocyte Society to standardize enrollment on clinical trials, HLH-94 and HLH-2004 (Table 1). Although extremely helpful as tools to capture the clinical and laboratory abnormalities characteristic of patients with HLH, the sensitivity and specificity of these criteria have not been prospectively validated in children or adults. Further, many features of HLH, such as progressive liver failure, disseminated intravascular coagulation, or encephalitis, are not captured. Regardless, the HLH-2004 criteria (Table 1) now serve as a de facto definition for HLH.3
Patterns of HLH
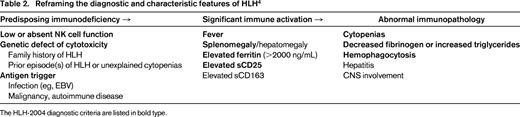
It is becoming increasingly clear that HLH represents a wide spectrum of clinical conditions that collectively arrive at the common endpoint of extreme, pathologic inflammation. Jordan et al have proposed a helpful framework to further conceptualize HLH in which a susceptible host with intrinsic immune defects experiences significant immune activation with resulting immunopathology (Table 2).4 Although episodes of immune activation are a universal human experience, physiologic mechanisms normally dampen the immune response prior to evolution of end-organ damage.
Primary and secondary HLH
When discussing HLH, patients are often declared to have primary or secondary HLH. “Primary” describes infants with predisposing inherited immune deficiencies, and “acquired” or “secondary” describes older patients in whom significant immune activation may be caused by a variety of antigen challenges, including autoimmune disease, persistent infection, malignancy, or loss of inhibitory immune mechanisms. These distinctions may be important to dissect the various mechanisms leading to HLH, but presumed categorization of primary/secondary in the acute setting is not clinically helpful. A systematic clinical investigation for immune triggers is essential for all HLH patients. This is especially true for adults with a new diagnosis of HLH in whom occult infection, malignancy or autoimmune disease are highly likely.5 Even patients with proven inherited HLH-associated gene defects generally require an infectious trigger to manifest symptoms.6 Therefore, coincident infection will not differentiate inherited from acquired disease. Outcomes on the HLH-94 trial were similar for patients with presumed primary and secondary HLH,7 indicating common risks of unbridled inflammation in all patients who arrive at this phenotype. With the exceptions of autoimmune and malignancy-associated HLH, initial evaluation and management is uniform for all patients. Identifying the presence of a fixed immune defect and risks of recurrence becomes more important later in management when determining need for continued immune suppression or hematopoietic stem cell transplant.
Host factors in HLH
The HLH genes
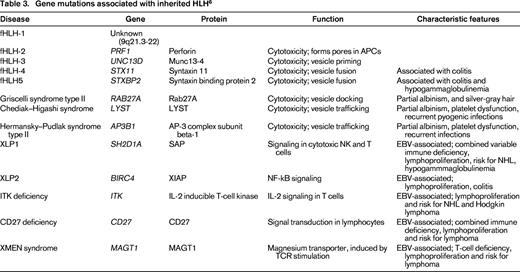
PRF1 (encoding perforin) was the first gene associated with inherited HLH.8 Subsequently, mutations in other genes have been characterized in patients with inherited HLH that regulate granule-dependent lymphocyte activity, including UNC13D (encoding Munc-13-4), STX11 (encoding syntaxin 11), and STXBP2 (encoding syntaxin-binding protein 2). Additional genes with function in intracellular granule trafficking affecting pigmentation as cytotoxic lymphocyte, neutrophil, and platelet function include RAB27A in Griscelli syndrome type II, LYST in Chediak–Higashi syndrome, and AP3B1 in Hermansky–Pudlak syndrome type II (for a review, see Jordan et al,4 Chandrakasan and Filipovich,6 and Janka and Lehmberg9 ). Genes that result in immune defects related to regulation of EBV reactivation are also associated with HLH, including SH2D1A and BIRC4/XIAP in X-linked lymphoproliferative disease (XLP1 and XLP2). More recently, mutations involved in T-cell function including IL-2-inducible T-cell kinase (ITK), CD27, and magnesium transporter gene (MAGT1; causes XMEN disease) have also been reported in association with EBV infection and HLH. Approximately one-half of children with HLH have identified gene defects, with wide variability based on region and ethnicity.4,6,10 Clearly, the list of “HLH-associated genes” (Table 3) will continue to grow.
Genotype and phenotype in HLH
Emerging data suggest that rather than the dichotomous “primary” or “secondary” categories, HLH may be more appropriately envisioned on a spectrum considering both inherited gene defects and polymorphisms of the host as well as specific immune challenges. Genotype/phenotype studies demonstrate that the in vitro impact of mutations on cytotoxic function correlates with age of disease onset. In general, mutations resulting in nonfunctional protein are associated with early disease,11-13 and hypomorphic mutations may present with first symptoms in adulthood. One large institutional series identified potentially functional mutations or polymorphisms in 14% of adults with clinical diagnosis of HLH, including patients >70 years of age.14 Further, digenic inheritance (single mutated allele from 2 HLH-associated genes) has also been observed, with single-allele inheritance of 2 degranulation genes (Rab27a, UNC13D, STXBP2, STX11) presenting in childhood and patients with a mutated degranulation allele and a defective PRF1 allele with later onset.15
Immune challenges and HLH
A vast array of immune challenges are associated with HLH including infection, malignancy, and autoimmune disease. Although improved sequencing technology is revealing an increasing number of genes associated with HLH, the impact of specific risk factors on development of HLH remain uncertain. Infection, autoimmune disease, and malignancy are relatively common events, rarely resulting in HLH. The gene defect with the most severe impact on cytotoxic lymphocyte function, ablated PRF1, almost universally result in HLH during infancy.11,14 On the other end of the spectrum, some viral illnesses, such as acute Ebola or Crimean–Congo Hemorrhagic Fever, induce a HLH-like phenotype in otherwise normal hosts.16,17 For the vast majority of HLH patients, disease likely results from complex interactions of host genetics and extrinsic immune challenges.
Infection
Innumerable case studies and series have reported particular viral, bacterial, fungal, and parasitic infections associated with HLH.18 Infection may represent an infectious trigger and/or an opportunistic event in a host with impaired immunity. EBV is the most frequent infection associated with HLH. It may arise as primary infection in any patient form of familial disease, and many without identified gene defects. Patients with predisposing immune deficiencies (eg, XLP or XMEN) are at especially high risk. Latent EBV, as well as other herpesviruses, may also reactivate due to impaired cytotoxic NK and T-cell function. In the setting of HLH, ethnic background influences propensity of EBV to infect T cells versus the normal B-cell target. Both circumstances can lead to potentially fatal acute lymphoproliferation or progression to severe chronic EBV (SCAEBV).19 A recent case report identified compound heterozygous mutations in PRF1 and STXBP2 in an adult with prolonged B-cell SCAEBV,20 consistent with inability to control EBV infection with impaired cytotoxic lymphocyte function. In a meta-analysis of adult HLH, EBV, HIV, and CMV were the most common viral infections; Mycobacterium tuberculosis and Rickettsia were the most common bacterial infections; Leishmania was the most common parasitic infection; and Histoplasma was the most common fungal infection.5
Distinctions between severe sepsis/SIRS and HLH remain blurred. The need for urgent initiation of therapy in HLH makes management of these infections in critically ill patients is challenging, as immune suppression may seem contraindicated. However, the importance of early control of pathologic inflammation initiation, along with therapy directed against infection, is supported by reports of delayed initiation of etoposide as a significant risk factor for early death in a multi-institutional study of adult HLH21 and in a national survey of children treated for EBV-associated HLH.22
Malignancy
HLH arises in the setting of many malignancies, most commonly associated with hematologic malignancies and often associated with coincident infections.23,24 Malignancy is the most common trigger identified in adults with HLH (45%), whereas malignancy is a significant but relatively rare event in pediatric HLH (8%).23,24 The causes of malignancy-associated HLH are variable and may include direct immune activation by transformed lymphocytes and/or loss of inhibitory immune function with development of disease-induced or treatment-induced bone marrow dysfunction. One study noted a high sIL2Rα/ferritin ratio was indicative of risk of lymphoma due to selective activation of transformed lymphocytes relative to benign macrophages.25 Many HLH-associated genes are also associated with increased risks of malignancy. Therefore, “malignancy-associated” HLH should not preclude a complete genetic evaluation. HLH may also arise in patients following hematopoietic stem cell transplant, likely because of combinations of selective immune reconstitution, infection, or graft versus host disease.4
Macrophage activation syndrome
There remains debate regarding the clinical and laboratory parameters that define MAS,26 but in general this designation is used to describe extreme inflammation in the setting of autoimmune disease, most commonly juvenile idiopathic arthritis (JIA) in children and systemic lupus erythematosus in adults.5,27 Continuing with the theme of blurring distinctions between presumed primary and secondary HLH, genomic studies of patients with JIA-associated MAS identified increased frequency of heterozygous functionally-significant mutations/polymorphisms in HLH-associated genes.28,29 Patients with MAS generally have depressed NK cell function, depressed perforin expression, and elevated sIL2Rα and CD163.30 Given the significant overlap between some extreme presentation of rheumatologic conditions such as Kawasaki disease or JIA, it is likely that the difference between “MAS” and “HLH” in some cases may depend on whether rheumatology or hematology is consulted first. In other cases, the distinction is important as patients with hyperinflammation/JIA because of causes other than impaired cytotoxic lymphocyte function may benefit from specific therapies, such as antagonism of interleukin-1 or interleukin-6.31
Synthetic HLH
The most recent addition to the hemophagocytic syndromes is iatrogenic. Several recent studies using chimeric antigen receptor-modified (CAR) T cells, bispecific T-cell engagers and cytotoxic T cells for cancer immunotherapy, have noted induction of a clinically-significant “cytokine release syndrome” associated with an HLH-like pattern of elevated ferritin, sIL2Rα, IFN-γ and IL-6, IL-8, and IL-10.32-34 Clinical symptoms may respond to treatment with tocilizumab, an IL-6-receptor inhibitor.33,34
Paths to pathologic hyperinflammation
Genotype and phenotype
Based on human studies, absence of PRF1 predictably results in HLH during infancy. However, it remains uncertain if PRF1 loss alone is sufficient to induce HLH without an immune trigger, as most infants are exposed to a large number of vaccines and infections during the first year of life. As discussed above, the majority of patients with HLH present with infections that could represent immune challenges and/or result from dysfunctional immunity. The first HLH mouse model with defective PRF1 required LCMV infection to induce the HLH phenotype.35 In fact, all mouse models of HLH with defects in granule-dependent cytotoxic lymphocyte function require specific infections to manifest the HLH phenotype, with severity of defects significantly associated with outcome. Further, severity in mouse models paralleled age of onset in human disease (PRF1>Rab27a>STX11>Lyst>HPS2).36 However, these single gene defects do not account for all mechanisms that underlie HLH. Mouse strains have particular sensitivity to manifesting the HLH phenotype. Presumably, genetic background in humans adds an additional level of mechanistic complexity as well.
Mechanisms of amplified inflammation
A critical clinical question that remains largely unanswered is how HLH is different from (or the same as) severe sepsis. Currently these diagnoses carry significantly different clinical approaches. In HLH, activated but unarmed lymphocytes are recruited to antigen presenting cells. Somehow, this then results in highly elevated proinflammatory cytokines in circulation (IFNγ, TNFα, IL-6, and M-CSF) and systemically activated lymphocytes and macrophages (Table 2, column 2). NK and cytotoxic T-cell dysfunction is fixed in “primary” HLH, and is also observed as a permanent or transient feature of many patients with “secondary” HLH.
Several mechanisms have been proposed to account for the explosive inflammation that results from impaired cytotoxicity.9 In mice, IFNγ production by CD8+ T cells is essential to recapitulate the HLH phenotype.37 An institutional series also identified higher IFNγ-related proteins in plasma of patients with HLH compared to other inflammatory conditions.38 One model suggests that activated CD8+ T cells normally prune a critical population of APCs that express infection-related antigens, driving continued T-cell activation and expansion. Persistent presentation, therefore, results in unchecked T-cell expansion and activation, ultimately manifesting as HLH.39 This model is consistent with the noted therapeutic efficacy of etoposide, an agent with enhanced toxicity against activated T cells.40 Although IFN-γ–mediated inflammation is a significant feature of HLH, the spectrum of mechanisms that potentially lead to pathologic inflammation are likely complex beyond a single pathway. Interestingly, persistent TLR9 signaling, a feature observed in human JIA, has also been shown to induce pathologic hyperinflammation in wild-type mice infected with LCMV.41,42 A conceptual framework for the range of mechanisms that may result in pathologic inflammation in HLH is presented in Figure 1.

Conceptual framework for mechanisms of HLH. HLH represents a group of syndromes in which pathologic inflammation results from a combination of immune dysfunction and immune activation. The contributions of inherited and acquired immune defects relative to the role of antigen stimulation may vary with age. For example, an infant in the NICU with PRF1 mutations with first upper respiratory infection would fall on the left, whereas an adult with HLH arising from persistent activation of transformed T cells in subcutaneous panniculitis-like T-cell lymphoma would fall on the right.
Conceptual framework for mechanisms of HLH. HLH represents a group of syndromes in which pathologic inflammation results from a combination of immune dysfunction and immune activation. The contributions of inherited and acquired immune defects relative to the role of antigen stimulation may vary with age. For example, an infant in the NICU with PRF1 mutations with first upper respiratory infection would fall on the left, whereas an adult with HLH arising from persistent activation of transformed T cells in subcutaneous panniculitis-like T-cell lymphoma would fall on the right.
Epidemiology of HLH
Defining the true incidence is an impossible task as HLH is a condition that some consider a faith-based diagnosis, making the phenotype of the provider as important as the patient to identify and report “HLH” versus other conditions characterized by inflammation.43 We acknowledge that the “I know it when I see it” rationale does not rise to modern medical diagnostic standards. However, there are some data that suggest the degree of inflammation described by the HLH-2004 criteria define a unique clinical group. First, the phenotype is severe in excess of most other inflammatory conditions: prior to immunochemotherapeutic treatment strategies, survival with patients diagnosed with HLH was <10%1 ; on HLH-94, 5 year overall survival was 54%, with 94% of deaths occurring in the first 8 weeks in patients with uncontrolled disease.7 These outcomes suggest “HLH” patients represent a group with critical pathologic inflammation that requires prompt immune suppression. Second, with improved understanding of genetic bases of inherited HLH and modern sequencing technology, every infant diagnosed in Sweden by the HLH-2004 criteria over a 6 year period had identified HLH-associated genetic mutations.44 Larger series estimate 40%-80% of patients diagnosed with HLH ultimately have gene-proven inherited disease.4 However imperfect, the HLH-2004 criteria confer some degree of specificity in identifying patients among the critically ill who merit consideration for diagnosis and treatment of HLH.
The Swedish national registry provides the a rigorous estimate the incidence of primary HLH with 1.5 cases/million live births in Sweden 2007-2011, up slightly from 1.2 cases/million in previous studies (1987-1996, 1997-2006).45 In North America, frequency of specific gene defects varies significantly with ethnicity/race.4 Genetic heterogeneity, presence of founder mutations, ethnic background, and frequency of HLH-associated triggers likely impact the local incidence of HLH in children and adults. At Texas Children's Hospital, we estimated diagnosis of HLH in 1/3000 inpatient admissions.46 We suspect that screening for and consideration of HLH also varies considerably based on level of suspicion and medical culture of individual institutions. Either the incidence of HLH in adults is exploding, or there is increased recognition: Pubmed search for “adult” and “hemophagocytic lymphohistiocytosis” in 2003-2004 identifies 17 publications, and in 2013-2014 identifies 206.
What is HLH?
As discussed above, a major challenge in discussing “HLH” is developing consensus on definition of the syndrome/disease. Clinical data provide strong support for the notion that patients meeting criteria are at very high risk for mortality from disease,7 and that outcomes have improved with increased recognition and immunochemotherapy in children.9,45 However, approaches to HLH currently remain relatively binary: a patient has or does not have HLH; based on diagnosis they receive HLH-94 (or similar) or supportive care. The debates around characterization and treatment of patients with MAS represent the personalized direction the field is heading. The “hemophagocytosis” in the disease name has led to some confusion regarding HLH as fixed biopsy-proven diagnosis like leukemia, where if there are blasts, then a specific protocol follows. Rather than focusing on adding up the diagnostic criteria for HLH as the goal of diagnostic evaluation, it should be the starting point. We propose a shift in emphasis to determine “pathologic inflammation due to X” (X = inherited or acquired immune dysfunction; autoimmune disease; malignancy; specific infection; unknown; or combination). By expanding the concept of HLH, there may be more room to develop graded or individualized approaches to diagnosis and therapy including consideration of the underlying immune disorder, specific antigen challenges, the “host,” as well as disease severity.
Correspondence
Carl E. Allen, Texas Children's Cancer Center, Texas Children's Hospital, 1102 Bates St, Feigin Center 730.06, Houston, TX 77030; Phone: 832-824-4312; Fax: 832-825-1206; e-mail: ceallen@txch.org.