Abstract
“Monoclonal gammopathy of clinical significance” (MGCS) is the term used to describe nonmalignant monoclonal gammopathies causing important disease. MGCS is the differential diagnosis for any patient presenting with what appears to be a monoclonal gammopathy of undetermined significance but is also experiencing other unexplained symptoms. Broadly, these conditions can be separated into symptoms and signs referable to the nerves, the kidneys, and the skin. The first step in making these diagnoses is to consider them. With a particular condition in mind, the next step is to order those tests that can help confirm or dismiss a particular diagnosis. Nearly all of the renal and dermatologic conditions are diagnosed by renal and skin biopsies, respectively. The importance of a highly competent renal pathologist and dermatopathologist cannot be underestimated. Biopsy is less specific for the neuropathic conditions. Because several of the MGCSs are syndromes, recognizing other manifestations is also key. Treatment recommendations for many of these conditions are anecdotal because of their rarity, but for several of the conditions, IV immunoglobulin, rituximab, and plasma cell–directed therapy are the best options.
Learning Objectives
• Recognize the differential diagnosis for a monoclonal gammopathy with peripheral neuropathy, skin abnormalities, or renal disease
• Diagnose MGCS
Clinical case
Approximately 18 months before diagnosis and at age 67, a man began experiencing anorexia, nausea, vomiting, and weight loss. By 12 months before diagnosis, he had lost 25 kg. An extensive nondiagnostic evaluation was performed, including thyrotropin, antineutrophil cytoplasmic autoantibodies, antinuclear antibodies, endoscopies, gastric emptying studies, and positron emission tomography–computed tomography. Seven months before diagnosis, an immunoglobulin Aλ monoclonal gammopathy was found. Over the ensuing months, this patient developed sensorimotor peripheral neuropathy, progressive muscle weakness, volume overload, and pseudogout. He was admitted to our hospital, where a diagnosis was made. In addition to the preceding presentations, he had malnutrition and anasarca (Table 1). His Eastern Cooperative Oncology Group performance status was 4.
Introduction
The term “monoclonal gammopathy of clinical significance” (MGCS) was coined subsequent to monoclonal gammopathy of renal significance (MGRS) when it became increasingly apparent that a term was required for a patient with a small B-cell clone and small monoclonal proteins that were causing serious and even life-threatening disease (Figure 1A).1,2 Monoclonal gammopathy of “undetermined significance” (MGUS) is a misnomer for these conditions. Some patients with MGCS have sufficient clonal burden to satisfy the definition of smoldering multiple myeloma or smoldering Waldenstrom macroglobulinemia. In short, MGCS is a monoclonal gammopathy featuring two main characteristics: a quiescent underlying clone and symptoms that are related to the monoclonal immunoglobulin or to the clone itself by mechanisms other than the tumor burden. The MGCSs are best divided into different systems that are affected, the most common of which are kidney, nerve, and skin, recognizing that in some cases there is overlap due to a systemic, multiorgan presentation and/or course.

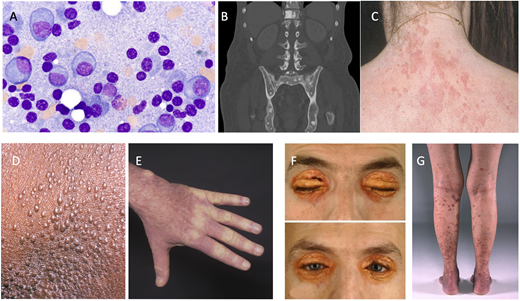
Part of the monoclonal gammopathy of clinical significance spectrum. (A) Bone marrow plasma cells (original magnification, 100×). (B) Osteosclerotic lesions of POEMS syndrome (polyradiculoneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes). (C) Schnitzler syndrome. (D and E) Scleromyxedema. (F) Necrobiotic xanthogranuloma. (G) Cryoglobulinemia vasculitis.
Part of the monoclonal gammopathy of clinical significance spectrum. (A) Bone marrow plasma cells (original magnification, 100×). (B) Osteosclerotic lesions of POEMS syndrome (polyradiculoneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes). (C) Schnitzler syndrome. (D and E) Scleromyxedema. (F) Necrobiotic xanthogranuloma. (G) Cryoglobulinemia vasculitis.
Neurologic MGCS
The major MGCS considerations for a patient with neuropathy include (Figure 2) amyloid light-chain (AL) amyloidosis, POEMS syndrome (polyradiculoneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes), cryoglobulinemia, CANOMAD (chronic ataxic neuropathy, ophthalmoplegia, immunoglobulin M [IgM] paraprotein, cold agglutinins, and disialosyl antibodies), and DADS-M (distal acquired demyelinating symmetric neuropathy with M protein [formerly known as “MGUS-associated peripheral neuropathy”]). The first three are diseases with multiple systemic manifestations, whereas the last two are primarily in the nervous system only.
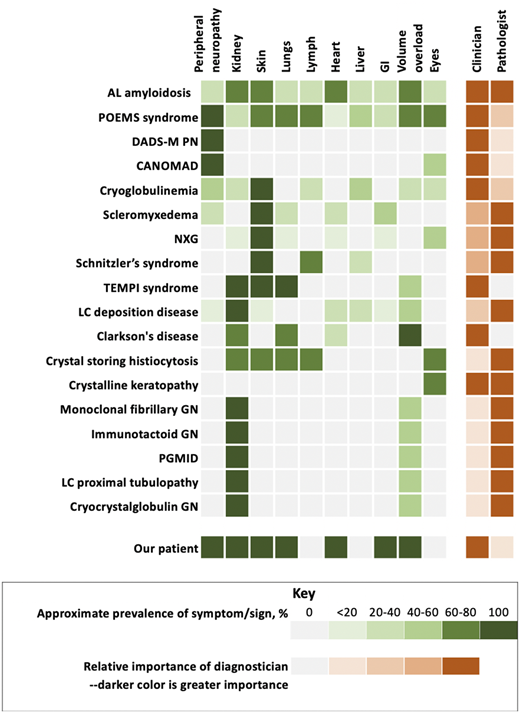
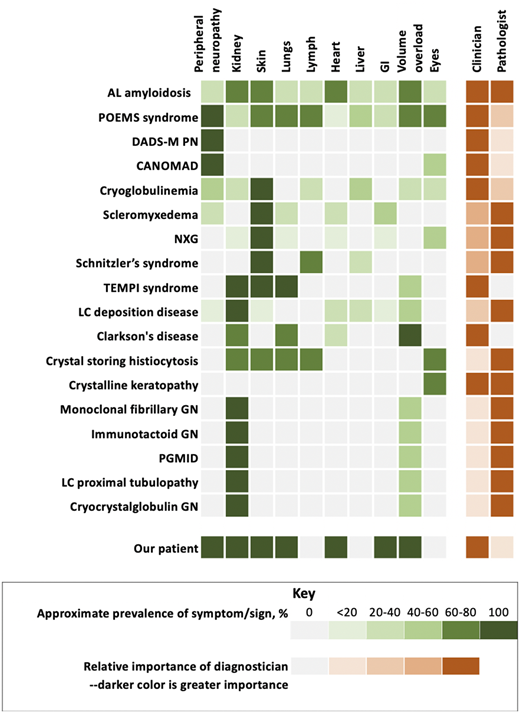
Monoclonal gammopathy of clinical significance and organ system involvement. CANOMAD, chronic ataxic neuropathy, ophthalmoplegia, immunoglobulin M paraprotein, cold agglutinins, and disialosyl antibodies; DADS-M-PN, distal acquired demyelinating symmetric neuropathy with M protein; GN, glomerulonephritis; LC, light chain; NXG, necrobiotic xanthogranuloma; PGMID, proliferative glomerulonephritis with monoclonal immune deposition; POEMS, polyradiculoneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes.
Monoclonal gammopathy of clinical significance and organ system involvement. CANOMAD, chronic ataxic neuropathy, ophthalmoplegia, immunoglobulin M paraprotein, cold agglutinins, and disialosyl antibodies; DADS-M-PN, distal acquired demyelinating symmetric neuropathy with M protein; GN, glomerulonephritis; LC, light chain; NXG, necrobiotic xanthogranuloma; PGMID, proliferative glomerulonephritis with monoclonal immune deposition; POEMS, polyradiculoneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes.
POEMS syndrome
POEMS syndrome is the acronym for polyradiculoneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes.3 It is a rare condition with a prevalence of 3 per 1 million. Median age at presentation is in the 50s, and there are slightly more men than women affected. Symptoms and signs refer to the acronym as well as features covered by another acronym, PEST, which includes papilledema, extravascular volume overload, sclerotic bone lesions (Figure 1B), thrombocytosis, and erythrocytosis. Other elements not covered in either of the acronyms are elevated vascular endothelial growth factor, pulmonary hypertension, reduced diffusion capacity of carbon monoxide, and arterial and venous thromboembolisms (Table 2). The dominant symptom in this disease is a progressive length-dependent ascending sensorimotor demyelinating peripheral neuropathy.
Adverse risk factors in this disease are age, pleural effusion, reduced estimated glomerular filtration rate, pulmonary hypertension, coexisting Castleman disease, and lack of complete hematologic response to plasma cell–directed therapy.4 Autologous stem cell transplant (ASCT) is a favored therapy, but lenalidomide and dexamethasone are also active (Table 3).5,6 Data associated with use of proteasome inhibitors and daratumumab are emerging. Overall survival in patients with POEMS syndrome is excellent with plasma cell–directed therapy, with estimated 10-year survivorship at 79%.
DADS-M
IgM monoclonal gammopathy accounts for ∼60% of neuropathies associated with monoclonal gammopathy.7 Patients are more often male and in their 50s to 80s. They present with a distal, demyelinating symmetric neuropathy. Sensory ataxia is the most common sign. The diagnosis is one of exclusion. Even in the presence of a monoclonal gammopathy, other explanations, such as inherited neuropathies, diabetes, alcoholism, and drug use, should be ruled out.
Pathologic studies have identified demyelination and widened myelin lamellae with IgM deposits in the widened lamellae of myelin fibers and myelin debris contained in Schwann cells and macrophages. Despite the fact that these M proteins may bind to myelin-associated glycoprotein (MAG) or other gangliosides, anti-MAG antibodies are not specific for peripheral neuropathy, and reduction in anti-MAG antibody titers with rituximab or other anti-CD20 antibodies has not correlated with clinical improvement. Treatments include IV immunoglobulin (IVIG) and rituximab.
CANOMAD
CANOMAD is a rare condition characterized by a chronic neuropathy with sensory ataxia and IgM disialosyl antibodies.8 Patients may or may not have motor weakness involving oculomotor and bulbar muscles or cold agglutinins. The most frequently targeted gangliosides are CD1b, GD3, GT1b, and GQ1b. Both axonal and demyelinating patterns have been recognized. The most effective therapies are IVIG, rituximab, and plasmapheresis.
Sporadic late-onset nemaline myopathy
Sporadic late-onset nemaline myopathy is a rare muscle disease that can be associated with a monoclonal protein or HIV infection.9 It is not a neuropathy, but it does cause significant motor dysfunction. On biopsy, muscle fibers accumulate nemaline rods, and there is no associated inflammation. Patients present with predominantly proximal or axial muscle weakness, including respiratory muscle weakness. Treatment strategies include IVIG and plasma cell–directed therapies, including ASCT.
Our patient had a symmetrical, ascending, demyelinating, sensorimotor peripheral neuropathy, along with systemic signs. The multisystemic nature of his disease made CANOMAD and DADS-M unlikely. Although AL amyloidosis and cryoglobulinemia are multisystemic diseases, the former is associated with a small-fiber neuropathy and the latter with an axonal neuropathy due to vasculitis.
Monoclonal gammopathy of renal significance
MGRS is a group of disorders in which a monoclonal immunoglobulin secreted by a nonmalignant or premalignant B-cell or plasma cell clone causes renal damage. As shown in Figure 2, several conditions are renal only, whereas others potentially have systemic features. All MGRS diagnoses without other systemic features are made by a renal pathologist (Figure 2). With the exception of C3 glomerulopathy with monoclonal gammopathy and thrombotic microangiopathy, the other MGRSs are broken into “nonorganized monoclonal immunoglobulin deposits” and “organized” monoclonal immunoglobulin deposits.10 The nonorganized deposits include monoclonal immunoglobulin deposition disease and proliferative glomerulonephritis with monoclonal immune deposits. In contrast, the organized deposits are categorized as (1) fibrillar deposits, which include AL amyloidosis and monoclonal fibrillary glomerulonephritis; (2) microtubular deposits, which include immunotactoid glomerulonephritis and cryoglobulinemia glomerulonephritis; and (3) inclusions or crystalline deposits, which include light chain proximal tubulopathy, crystal-storing histiocytosis, and cryocrystalglobulin glomerulonephritis.
This classification allows for a uniform vocabulary among nephrologists, renal pathologists, and hematologists such that diagnostic algorithms can be constructed and natural history and therapeutic interventions can be analyzed (Table 3). Although renal transplant can be considered in these patients, there is risk for recurrence.11 Which diseases respond best to which plasma cell or lymphoproliferative disease–directed therapies is still a work in progress. The most data exist on light chain deposition disease (LCDD), so that topic is described next.
LCDD
LCDD shares similarities with AL amyloidosis in that both are immunoglobulin deposition diseases. Both can rarely involve immunoglobulin heavy chains as well. LCDD is less common than AL amyloidosis, less likely to involve other organs, and more often is due to a κ-restricted light chain (immunoglobulin kappa variable 4 [IGKV4]) and to present with impaired creatinine clearance. The median plasmacytosis in the bone marrow is ∼10%.12 ,13 Immunofluorescence of the kidney biopsy reveals linear deposits of the involved monoclonal immunoglobulin along tubular basement membranes and along glomerular basement membranes. Electron microscopy shows that these deposits have a granular appearance.
Treatment is similar to that used for patients with AL, and survival tends to be better because it is unusual for patients with LCCD to have cardiac involvement. Although our patient had reduced renal function and mild proteinuria, no renal biopsy was performed. The renal injury was presumed to be due to hypotension and exuberant application of diuretics. Significant renal disease is rare in POEMS syndrome, but numerous pathologic changes have been reported, including membranoproliferative glomerulonephritis–like lesions, microangiopathic lesions, mesangiolytic lesions, and even immunotactoid lesions.
Cutaneous MGCSs
Cutaneous MGCSs include Schnitzler syndrome, scleromyxedema, necrobiotic xanthogranuloma (NXG), TEMPI syndrome (telangiectasias, elevated erythropoietin and erythropoiesis, monoclonal gammopathy, perinephric fluid, intrapulmonary shunting), cryoglobulinemia, systemic capillary leak syndrome (SCLS), and POEMS syndrome.
Schnitzler syndrome
Schnitzler syndrome is characterized primarily by chronic urticaria (Figure 1C) and the presence of an IgM monoclonal gammopathy. Interleukin (IL)-1β plays a critical role in in the disease. Aberrant NLRP3 inflammasome signaling and cytokine pathway dysregulation also play a role. Schnitzler syndrome can rarely be associated with an IgG monoclonal gammopathy. Other features and diagnostic criteria are shown in Figure 2 and Table 2.14-16 Although the presence of a dermal neutrophilic infiltrate on skin biopsy became a minor criterion with the Strasbourg revision, such a biopsy finding is nonspecific, and its relative importance in the diagnostic criteria has been questioned. Therapy with anakinra, an anti–IL-1 antibody, is quite effective (Table 3). More novel anti–IL-1 antibodies, such as rilonacept and canakinumab, have also been effective.17 Following C-reactive protein can be helpful in monitoring this disease.
Scleromyxedema
Scleromyxedema is characterized by generalized papular and sclerodermoid cutaneous eruptions (Figure 1D-E) and is typically associated with an IgG monoclonal gammopathy.18 Extracutaneous involvement can include the nervous system, joints, gastrointestinal system, and heart (Figure 2). The infiltrates are composed of mucin. How the monoclonal protein induces fibroblast proliferation is not well understood. The science behind the pathology is gradually emerging through skin transcriptome analyses and the study of peripheral blood immune cells. Transforming growth factor-β is overexpressed. Other proteins, including collagen Ia and several interferon-inducible proteins, are also overexpressed.19,20 Diagnostic criteria are shown in Table 2. IVIG is considered first-line therapy, with plasma cell–directed therapy (eg, lenalidomide or bortezomib) added if no response is achieved or more severe disease develops (Table 3).20
NXG
NXG is a non–Langerhans cell histiocytosis typically associated with monoclonal proteins attributable to plasma cell disorders or lymphoproliferative disorders. The mean age of presentation is 62 years, with a slight predominance of women.21 The classic presentation is yellow-to-orange papules, plaques, and/or nodules involving the eyelids (Figure 1F). Cutaneous lesions may also be found on other locations of the face, the trunk, and the extremities. NXG plaques can occasionally be pruritic and also painful if they ulcerate. Extracutaneous involvement includes the eye, heart, gastrointestinal tract, liver, and lung but is relatively rare (Figure 2). A French group has suggested an association between monoclonal gammopathy and both hyperlipidemic and nonhyperlipidemic xanthomatosis and suggested that the association can be strengthened if complement levels, especially C4, are low.22
On biopsy of NXG, palisading granulomas with nonclonal lymphoplasmacytic infiltrate and zones of necrobiosis are seen. Cholesterol clefts and large bizarre foreign body giant cells are also classic. The pathogenesis of the disease is unknown, but it has been speculated that there is a monoclonal protein–lipoprotein interaction. Diagnostic criteria have been proposed (Table 2). The leading differential diagnosis is necrobiosis lipoidica, which is a necrotizing skin condition that can occur in patients with diabetes mellitus or rheumatoid arthritis. In contrast to monoclonal gammopathy–associated hyperlipidemic and nonhyperlipidemic xanthomatosis, CD163-positive foam cells and Touton giant cells are seen but typically without necrobiosis. Treatment with IVIG is one of the most promising therapies, but limited success has also been reported with plasma cell–directed therapies, intralesional triamcinolone, and antimalarials.
TEMPI syndrome
TEMPI syndrome is a rare acquired disorder characterized by the features that comprise the acronym: telangiectasias, elevated erythropoietin and erythrocytosis, monoclonal gammopathy, perinephric fluid collections, and intrapulmonary shunting.23 The underlying pathophysiology is not understood, but it is clear that plasma cell–directed therapy reverses the clinical manifestations.
Telangiectasias involving the face and upper body and erythrocytosis comprise the most common presentation. Unlike the erythrocytosis of polycythemia rubra vera and of POEMS syndrome, patients with TEMPI syndrome have a high erythropoietin level.24 These patients develop progressive hypoxia. The pulmonary shunting is not evident on high-resolution computed tomography of the chest and is best demonstrated by 99mTc macroaggregated albumin scintigraphy. The perinephric fluid collections have the same electrolyte composition as serum. Proposed diagnostic criteria are shown in Table 2. Unlike POEMS syndrome, there is no bias in clonality for λ-restricted clones, and there are no features of a myeloproliferative neoplasm.
Plasma cell–directed therapy appears to be useful, specifically bortezomib, daratumumab, lenalidomide, and high-dose melphalan. All of the features can improve upon achievement of complete hematologic response (Table 3).
Cryoglobulinemia
Cryoglobulinemia is a multisystem disease that can affect almost any organ system, but cutaneous manifestations are almost always present (Figure 1G). Type I cryoglobulins arise from clonal plasma cell proliferative disorders or lymphoproliferative disorders. Type II and type III cryoglobulins may be related to plasma cell disorders or lymphoporliferative disorders but are more often due to infections such as hepatitis C virus and connective tissue disorders.25
Manifestations and disease severity are quite variable. Type I cryoglobulins more often cause occlusive symptoms due to occlusion of capillary lumina, and vasculitis is uncommon.25 Patients report cold-induced skin symptoms, including purpura, livedo, and cold urticaria. Ulceration can occur. Less than one-third of patients will have renal involvement, but up to 50% may have peripheral neuropathy. In contrast, in type II/III cryoglobulinemia or mixed cryoglobulinemia, small-vessel vasculitis is the major mechanism driving morbidity. Skin symptoms, including purpura, occur in the vast majority of patients; arthralgia is also very common in peripheral neuropathy followed by renal involvement.
The aim of treatment of cryoglobulinemia is to treat the underlying cause. For patients with hepatitis C virus, who comprise the majority of type II and III cases, sustained virologic responses can be achieved in >50% with antiviral therapy. For patients not responding to antiviral therapy, rituximab and other immunosuppressants can play an important role in treating vasculitis. For disease driven by autoimmune disease, rituximab and corticosteroids are the best first-line options. Plasmapheresis can be used in patients with severe end organ damage and/or refractory disease.26 Other disease modifiers, including corticosteroids and cyclophosphamide, can also play a role in therapy, especially in patients with severe end organ damage.
Idiopathic SCLS (Clarkson disease)
This devastating disease was first described in 1960. SCLS is characterized capillary leak resulting in sudden-onset shock and anasarca caused by plasma extravasation (up to 70% of total plasma volume). The diagnostic triad is composed of the “3 Hs,” which occur in the absence of secondary causes of these findings: hypotension, hemoconcentration, and hypoalbuminemia. Sixty-eight percent of adult patients with SCLS have monoclonal proteins, most commonly IgG-κ. Details of the limited understanding of disease mechanisms of the vascular endothelial hyperpermeability of SCLS can be found in a review published elsewhere.27
The differential diagnosis for an acute attack includes sepsis, anaphylaxis, and hereditary angioedema. Treatment at the time of an acute attack is supportive with fluid resuscitation until flare subsides, which typically occurs over the course of a few days. Empiric prophylaxis with IVIG is recommended because it has been demonstrated that there are fewer attacks in those patients managed as such.28
POEMS syndrome
Our patient had POEMS syndrome evolving over the course of nearly 18 months. His first symptoms were those of adrenal insufficiency and peripheral neuropathy. His characteristics and his course are summarized in Figure 2 and Table 1. His case exemplifies the importance of an extensive review of systems in patients with a monoclonal protein and other unexplained conditions. A focused review of systems prompts additional testing from scans to disease-oriented blood work and/or biopsies. By the time he was diagnosed, our patient had a performance score of 4 and was bedridden with bedsores. With adrenal and thyroid replacement and plasma cell clone-directed therapy, his life has returned to nearly normal, though he does still use ankle foot orthotics. Had the syndrome not been considered, he would have died. Five years later, he continues to enjoy life.
In summary, MGCSs are a constellation of diseases associated with clonal—but not malignant–B cells or plasma cells that produce monoclonal proteins and pathology through diverse, often ill-defined mechanisms. Similar end organ damage can occur in the context of malignant plasma cell or B-cell clonal disorders, but these are no longer MGCSs, but rather the cancer with associated disease (eg, myeloma with associated cryoglobulinemia). The most commonly affected organs among patients with MGCS are the kidney, nerve, and skin. A thorough discussion of all of the MGCSs is beyond the scope of this article, but Table 4 highlights some of the most recent and relevant references pertaining to MGCS. Some MGCSs predominantly affect only one organ, and others are systemic diseases affecting multiple organ systems. In order to help patients, these diagnoses and the severity of the symptoms must be considered so that appropriate therapy may be instituted. Conversely, not every patient with a monoclonal gammopathy and an unexplained symptom or sign has MGCS, given the high prevalence of true MGUS, especially with advancing age. In many instances, the appropriate therapy is clone-directed therapy, even though there is no malignancy; in others, data indicate that therapies such as IVIG, rituximab, or anti–IL-1 antibodies are most appropriate. The treating physician should strive to assign causality before proceeding with clone-directed therapy.
Correspondence
Angela Dispenzieri, Division of Hematology, Mayo Clinic, 200 First St, SW, Rochester, MN 55905; e-mail: dispenzieri.angela@mayo.edu.