Abstract
With our increasing understanding of inherited marrow failure and myeloid malignancy predisposition syndromes, it has become clear that there is a wide phenotypic spectrum and that these diseases must be considered in the differential diagnosis of both children and adults with unexplained defects in hematopoiesis. Moreover, these conditions are not as rare as previously believed and may present as aplastic anemia, myelodysplastic syndrome, or malignancy over a range of ages. Establishing the correct diagnosis is essential because it has implications for treatment, medical management, cancer screening, and family planning. Our goal is to highlight insights into the pathophysiology of these diseases, review cryptic presentations of these syndromes, and provide useful references for the practicing hematologist.
Learning Objectives
Review the basic clinical features of the inherited BM failure and myeloid malignancy predisposition syndromes that can lead to aplastic anemia, myelodysplasia, and malignancy
Emphasize the role of a thorough physical exam and detailed family history to identify these disorders and determine the need for genetic testing for the patient and family
Discuss the initial diagnostic workup of these disorders and potential treatment modifications if a specific condition is identified
Introduction: Inherited bone marrow failure syndromes
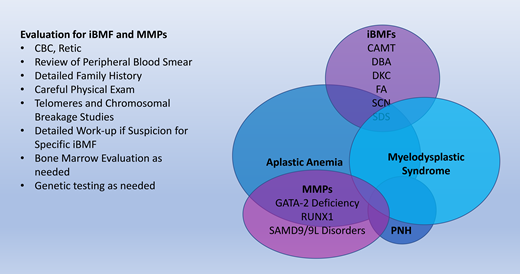
Inherited bone marrow failure syndromes (iBMFs) encompass a diverse collection of diseases. While they are rare causes of hematologic disorders, it is essential for the practicing hematologist to be aware of iBMFs. These disorders have a wide phenotypic spectrum and may present cryptically in adult patients with cytopenias in one or more lineages. Furthermore, our evolving understanding of the pathophysiology of these disorders is critical to our general knowledge of the hematopoietic system. An overview of these syndromes is summarized in Table 1. We have highlighted the disorders that have a predisposition to aplastic anemia, myelodysplastic syndrome (MDS), and malignancy. A brief overview of each of these disorders follows.
Congenital amegakaryocytic thrombocytopenia
Congenital amegakaryocytic thrombocytopenia (CAMT) is a rare autosomal recessive disease characterized by an isolated, severe hypomegakaryocytic thrombocytopenia that presents in infancy and progresses to pancytopenia and bone marrow (BM) failure in later childhood. Clinical manifestations can include petechiae at birth and intracranial hemorrhage, but unlike other iBMFs, congenital anomalies are rare. CAMT is most commonly due to mutations in MPL, which encodes for the thrombopoietin receptor. In contrast to the activating MPL mutations seen in myeloproliferative neoplasms, CAMT mutations always lead to diminished or absent signaling of the thrombopoietin receptor. In general, loss-of-function mutations are associated with more severe thrombocytopenia and early-onset pancytopenia (CAMT type 1). In cases with MPL gene missense mutations, patients have a transient increase in platelet counts during the first year of life (CAMT type 2). Heterozygous mutations in MECOM (MDS1 and EVI1 complex locus) have been reported to be causative of a rare association of CAMT and radioulnar synostosis. Mutations in MECOM can present with a wide range of phenotypes ranging from thrombocytopenia to full-blown hypocellular marrow failure/aplastic anemia early in life, again demonstrating the importance of screening for these rare diseases and the need for ongoing scientific discovery.
Diamond-Blackfan anemia
Diamond-Blackfan anemia (DBA) was originally described by Josephs in 1936 and further characterized by Diamond and Blackfan in 1938 as a congenital hypoplastic anemia.1 In addition to hypoplastic anemia, the disorder is characterized by macrocytosis, reticulocytopenia, and elevated levels of erythrocyte adenosine deaminase. Additionally, half of patients with DBA also present with physical abnormalities, including short stature, thumb abnormalities (classically, a triphalangeal thumb), craniofacial defects, and cleft lip/palate. DBA was the first disease to be linked to impaired ribosome function and is the founding member of a group of disorders now known as ribosomopathies.2 Several other iBMFs as well as the 5q− MDS and cancers have subsequently been linked to mutations in genes encoding for ribosomal proteins or proteins required for normal ribosome function.3,4 While the predisposition to MDS is lower than other iBMFs, there is a risk of other tumors, including colorectal cancer. In addition, DBA is known to have incomplete penetrance, as demonstrated both by the observation of siblings who carry the same mutation discordant for the presence of anemia and by the occurrence of spontaneous remissions in affected patients, which is why clinicians need to have a high index of suspicion in patients with unexplained anemia and marrow abnormalities.
Dyskeratosis congenita
In 1910 a case report was the first to define a syndrome, ultimately named dyskeratosis congenita (DC), characterized by a triad of abnormal skin pigmentation, oral leukoplakia, and nail dystrophy.5 The spectrum of DC has expanded considerably since then to include effects on every organ system, particularly the BM. Other clinical findings may include early graying of hair or hair loss, short stature, developmental delay, blepharitis, periodontal disease, pulmonary fibrosis, esophageal stenosis, urethral stenosis, liver disease, and avascular necrosis of the hips or shoulders. Almost 90% of patients with DC will eventually develop a cytopenia of one or more peripheral blood lineages, and BM failure is the major cause of death. Often BM failure develops in the second or third decade of life, but it can occur at birth or as late as the seventh decade of life. The BM abnormalities can evolve into MDS or acute myeloid leukemia (AML).6
The clinical diagnosis of DC can be challenging given its phenotypic heterogeneity, different modes of inheritance (X-linked, autosomal recessive, and autosomal dominant), and variable age of onset. However, despite the wide spectrum of the disease, ranging from classic DC to aplastic anemia, it is clear that the underlying pathology is due to defective telomere maintenance. The term “DC” is now used interchangeably with telomere biology disorders and short telomere syndromes. The functional assessment of telomere length with the finding of a telomere length less than the first percentile for age in leukocyte subsets is highly sensitive but not specific to the diagnosis of these disorders and has been seen in other inherited disorders.7,8 Regardless, the evaluation of telomere length is critical for any patient with pancytopenia and abnormal marrow findings given the implications for screening and modifications in stem cell transplant conditioning.
Fanconi anemia
Fanconi anemia (FA) was first described by Guido Fanconi in 1927 in 3 brothers with pancytopenia and congenital abnormalities.9 The disease typically progresses through several clinical stages, organized by age. In the first stage, in infancy and early childhood, congenital anomalies may be present, although they are not required for the diagnosis of FA and range from mild to severe. The most common malformations include short stature, hypopigmented or café au lait spots, thumb or radial ray abnormalities, micro- or hydrocephaly, structural renal anomalies, hypogonadism, and developmental delay.
Within the first decade, patients may present with thrombocytopenia and macrocytosis before progressing to BM failure, when the diagnosis is often first made. During adolescence and adulthood, the risk of AML and MDS becomes very high. In older adults, a range of solid tumors, particularly squamous cell carcinomas of the head/neck and genitourinary track, can be seen.10 Throughout life, the hematopoietic phenotype can change because of genetic reversion or clonal evolution, so a high index of suspicion and a careful history, including a family history of cancer predisposition, is essential. These patients are highly sensitive to chemotherapy and radiation, which is why it is critical to rule out this diagnosis prior to transplant for any patient with unexplained pancytopenia.
Severe congenital neutropenia
Severe congenital neutropenia (SCN) is characterized by absolute neutrophil counts consistently less than 200/uL with recurrent severe infections, which often develop within the first few months of life. Evaluation of the BM demonstrates a myeloid maturation arrest. The disease can be autosomal recessive (Kostmann syndrome), X-linked, or autosomal dominant. In Kostmann syndrome, mutations have been identified in the HAX1, G6PC3, and GFI1 genes. In the X-linked disease, mutations have been found in the WAS gene. Finally, mutations in the ELANE gene are the most common cause of SCN and cause autosomal dominant disease. As with Shwachman-Diamond syndrome, below), additional somatic mutations are often acquired over time, which serve as important markers for disease risk and may affect treatment decisions.
Shwachman-Diamond syndrome
Shwachman-Diamond syndrome (SDS, also known as Shwachman- Diamond-Oski syndrome) was initially described in the early 1960s as a disorder of exocrine pancreatic dysfunction and BM failure.11 Patients generally present with steatorrhea, growth failure, and recurrent infections. The hematologic abnormalities are predominantly neutropenia, although anemia, thrombocytopenia, and pancytopenia can occur. Skeletal abnormalities (short stature, delayed appearance of normally shaped epiphyses, and progressive metaphyseal thickening/dysplasia) as well as poor growth are common in SDS patients. Patients without severe pancreatic disease may present later in childhood or as adults, and some patients may present with aplastic anemia or AML as the initial manifestation of the disease.12
The diagnosis of SDS relies on clinical findings. Exocrine pancreatic dysfunction, which can be subclinical, can be established by low serum trypsinogen in patients younger than 3 years or low pancreatic isoamylase in patients older than 3 years. In addition, patients may have low fecal elastase or a fatty pancreas that can be demonstrated by imaging. Genetic testing can be helpful to confirm the diagnosis, but a negative test does not exclude the diagnosis. In 90% of affected patients, this autosomal recessive disorder is due to mutations in the Shwachman-Bodian-Diamond syndrome (SBDS) gene.13 It is essential to distinguish SDS from cystic fibrosis (the most common cause of exocrine pancreatic insufficiency in children), congenital neutropenia, and Pearson syndrome.
Myeloid malignancy predisposition syndromes
GATA-2 deficiency
The constellation of disseminated nontuberculous mycobacterial infections; susceptibility to viral infections, especially human papillomavirus and Epstein-Barr virus; and fungal infections, coupled with profound monocytopenia and B-cell and natural killer cell lymphopenias, was syndromically described as MonoMAC and dendritic cell, monocyte, B, and natural killer lymphoid deficiency. Mutations in GATA-2 were identified in 2011 as the cause of this group of disorders, which are also associated with marrow failure, MDS, and AML.14 The disease follows an autosomal dominant mode of inheritance as well as a large number of sporadic cases. Presentations vary widely among affected members even within the same family, but the BM is often initially hypocellular and may have striking multilineage dysplasia.
RUNX1
RUNX1 is a member of the core-binding factor family of transcription factors and is indispensable for definitive hematopoiesis; it is one of the most frequently mutated genes in a variety of hematological malignancies. Germ line mutations in RUNX1 have been shown to cause a familial platelet disorder with associated malignancies, including pediatric MDS. Patients may present with isolated thrombocytopenia and megakaryocytic dysmorphia or atypia on baseline BM evaluation before progressing to pancytopenia, multilineage dysplasia, increased blasts, and the acquisition of additional somatic mutations. A subset of patients may present with MDS/AML at a young age. Early recognition of germ line mutation and predisposition to myeloid malignancy permits appropriate treatment, adequate monitoring for disease progression, and proper donor selection for hematopoietic stem cell transplantation, as well as genetic counseling of affected patients and their family members.
SAMD9/9L disorders
Germ line SAMD9 and SAMD9L mutations have now been identified as the cause of a spectrum of multisystem disorders that carry a markedly increased risk of developing myeloid malignancies with chromosome 7 abnormalities, including monosomy 7, del7q, and CN-loh of 7q. Affected individuals display a highly variable clinical course that ranges from mild and transient dyspoietic changes in the BM to a rapid progression of MDS or AML with monosomy 7. These data have implications for understanding how SAMD9 and SAMD9L mutations contribute to myeloid transformation and for recognizing, counseling, and treating affected families.
The role of genetic testing
Many of the disorders discussed have specific, identified gene mutations, but there is increasing interest in understanding what other genes may be involved within these diseases as well as in disorders in which no genes have been identified. Increasing numbers of panels are being conducted both in commercial labs and universities that cover BMF only vs whole exome sequencing. The decision about specific tests to be performed should be done in consultation with a geneticist, which will also help guide decisions about family testing. This information is critical for surveillance and may help steer treatment options regarding stem cell donor transplantation.
Acquired BM failure syndromes that can present with signs of marrow aplasia
Although beyond the scope of this chapter, it is worth noting that several genetic causes of acquired BM failure should be considered in the evaluation of patients with cytopenias and hypoplastic marrow. Again, a detailed physical exam and a careful family history will help to determine the appropriate workup and genetic testing. For example, VEXAS syndrome is a monogenic disease of adulthood caused by somatic mutations in UBA1 in hematopoietic progenitor cells that can present with a range of immunologic and hematologic symptoms.15
Conclusions
Maintaining a high suspicion for rare congenital and acquired causes of BM failure is critical when evaluating patients of all ages with unusual cytopenias. A detailed physical examination and family history are also critical. These guide the subsequent laboratory workup, surveillance schedule for malignancy, and potential therapeutic options.
Conflict-of-interest disclosure
Anupama Narla: no competing financial interests to declare.
Off-label drug use
Anupama Narla: nothing to disclose.