Abstract
Monoclonal gammopathies of clinical significance (MGCS) are a heterogeneous group of disorders characterized by the presence of an indolent B-cell or plasma-cell clone producing a toxic monoclonal immunoglobulin resulting in end-organ dysfunction. MGCS is a clinicopathologic diagnosis that requires the demonstration of a monoclonal immunoglobulin in the correct clinical setting. The most common MGCS syndromes are renal, neurologic, and cutaneous, although hematologic and multi-organ MGCS syndromes are also increasingly recognized. Therapy most commonly targets the underlying clonal population; immunoglobulin-targeting therapies as well as complement and cytokine antagonists have emerged for selected MGCS syndromes and may be temporizing in a subset of patients. Other chapters review renal and neurologic MGCS; this chapter focuses on hematologic and multi-organ MGCS syndromes.
Learning Objectives
Recognize MGCS as a distinct entity closely related to MGUS, multiple myeloma, Waldenström macroglobulinemia, and light chain amyloidosis
Develop a differential diagnosis for MGCS
Select appropriate therapies for hematologic and multi-organ MGCS syndromes
CLINICAL CASE
A 35-year-old woman presented with 2 years of intermittent painful, pruritic rash. Serum protein immunoelectrophoresis demonstrated an IgM kappa paraprotein at 0.1 g/dL, and serum free light chains were normal. Levels of complement factors C3 and C4 were low, and inflammatory markers were elevated. Renal function was normal and urine sediment was bland. A cryoprotein screen demonstrated a cryocrit of 10%, and immunofixation demonstrated monoclonal IgM kappa; rheumatoid factor was absent. Bone marrow examination revealed 5% involvement by a kappa-restricted plasma-cell clone; there was no B-cell component, and fluorescence in situ hybridization studies were negative. Steroids temporarily relieved pruritus but were poorly tolerated. Three months of weekly bortezomib resolved rash and cryoprotein. Four years later, the rash and cryoprotein returned, and a 6-month course of daratumumab was administered, with favorable treatment response to date.
Introduction
Monoclonal gammopathies are a heterogeneous group of disorders characterized by the presence of a monoclonal immunoglobulin in the serum and/or urine. Underlying these monoclonal proteins are clonal populations of B cells, plasma cells, or both.1 Qualitative factors, such as the clonal cell type and identity of the monoclonal paraprotein, as well as quantitative factors, such as the degree of marrow infiltration and concentration of the monoclonal immunoglobulin, largely dictate the clinical behavior of these disorders.
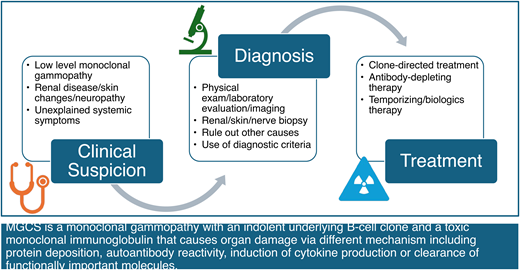
Monoclonal gammopathies of clinical significance occur when an otherwise indolent lymphoproliferative clone gives rise to end-organ dysfunction via paraprotein-mediated toxicity. Figure 1 illustrates the relationship between MGCS to other well-described B- and plasma-cell disorders, highlighting whether end-organ dysfunction is mediated predominantly by the clonal proliferation, by the paraprotein, or by both. There are several MGCS subtypes, classified by the dominant organ system involved, including monoclonal gammopathy of renal, neurologic, cutaneous, and hematologic significance; in addition, MGCS can present as a multi-organ syndrome (Figure 2).
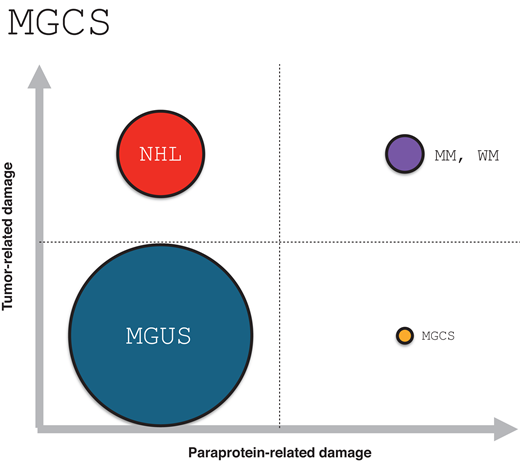
A simple schema illustrating the relationship between MGUS, MM and WM, NHL, and MGCS based on the dominant cause of end-organ dysfunction. Circle diameter reflects estimate of relative incidence (not to scale). MGCS, monoclonal gammopathy of clinical significance; MGUS, monoclonal gammopathy of undetermined significance; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; WM, Waldenström macroglobulinemia.
A simple schema illustrating the relationship between MGUS, MM and WM, NHL, and MGCS based on the dominant cause of end-organ dysfunction. Circle diameter reflects estimate of relative incidence (not to scale). MGCS, monoclonal gammopathy of clinical significance; MGUS, monoclonal gammopathy of undetermined significance; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; WM, Waldenström macroglobulinemia.

Overview of select subtypes of monoclonal gammopathy of clinical significance. acquired vWD, acquired von Willebrand disease; CAD, cold agglutinin disease; CANOMAD, chronic ataxic neuropathy, ophthalmoplegia, immunoglobulin M paraprotein, cold agglutinins, and disialosyl antibodies; DADS, distal acquired demyelinating symmetric neuropathy; MGRS, monoclonal gammopathy of renal significance; MGTS, monoclonal gammopathy of thrombotic significance; NXG, necrobiotic xanthogranuloma.
Overview of select subtypes of monoclonal gammopathy of clinical significance. acquired vWD, acquired von Willebrand disease; CAD, cold agglutinin disease; CANOMAD, chronic ataxic neuropathy, ophthalmoplegia, immunoglobulin M paraprotein, cold agglutinins, and disialosyl antibodies; DADS, distal acquired demyelinating symmetric neuropathy; MGRS, monoclonal gammopathy of renal significance; MGTS, monoclonal gammopathy of thrombotic significance; NXG, necrobiotic xanthogranuloma.
This chapter focuses on the group of MGCS associated with multi-organ syndromes and hematologic MGCS (Table 1). Other chapters are dedicated to renal and neurologic MGCS. We begin with a review of the diagnostic studies required to document the presence of monoclonal gammopathy.
Diagnostic evaluation
MGCS should be considered in a patient with monoclonal gammopathy and unexplained end-organ dysfunction plausibly related to the paraprotein. The diagnosis of monoclonal gammopathy deserves attention.
Diagnosing monoclonal gammopathy
Initial descriptions of monoclonal gammopathy hinged on the detection of an intact monoclonal immunoglobulin composed of heavy and light chains migrating as a homogeneous band on serum or urine protein electrophoresis. Most individuals with monoclonal gammopathy have monoclonal gammopathy of undetermined significance (MGUS), which, contrary to common wisdom, is not a laboratory diagnosis but rather a clinicopathologic diagnosis. In MGUS, the lymphoproliferative clone is indolent, and the associated monoclonal protein is essentially inert. A subset of patients with MGUS experience progression to symptomatic disease that warrants therapy, at a rate of approximately 1% per year, but the majority live an otherwise normal life without MGUS progression.2 Early screening studies indicated that MGUS occurs frequently in healthy individuals, with a prevalence of approximately 3% among individuals 50 years and older.3
With the introduction of the serum free light chain (FLC) assay came the observation that monoclonal gammopathy can also occur in the absence of an intact immunoglobulin, and the concept of FLC monoclonal gammopathy emerged.4 FLC monoclonal gammopathy occurs when excess monoclonal kappa or lambda FLC, unbound to an associated heavy chain, is detected in the serum or urine, along with a skewed ratio of kappa to lambda FLC.
Diagnosing FLC monoclonal gammopathy poses challenges, in part due to limitations of the serum FLC assay, in which the ratio of serum kappa to lambda FLC is a surrogate for rather than direct measurement of monoclonality. This surrogacy, and the fact that serum FLCs are routinely elevated in reactive states and from poor clearance (eg, inflammatory disorders and renal dysfunction), necessarily impairs the performance of the assay. Probably because of overdiagnosis, the rate of progression of FLC MGUS is threefold lower than that of intact immunoglobulin MGUS.4 Several efforts, including the development of high- resolution mass spectroscopy that can directly measure monoclonal FLC rather than relying on the surrogacy of a skewed serum FLC ratio, are poised to greatly enhance diagnostic accuracy among patients with suspected FLC monoclonal gammopathy.5 In the meantime, several initiatives have aimed at refining serum FLC reference ranges in patients with inflammatory disorders,6 in those with renal dysfunction,7 and in heathy individuals undergoing population-level screening.8 Recently proposed reference ranges for serum FLCs are highlighted in Table 2.
Differentiating between MGUS and MGCS
Once monoclonal gammopathy is confirmed in a patient with an unexplained syndrome, attention is turned to demonstrating a causal association between the paraprotein and the end- organ dysfunction. It is important to reiterate that the frequency of MGUS implies that a large subset of patients in whom MGCS is considered will in fact have MGUS after the diagnostic evaluation is complete.
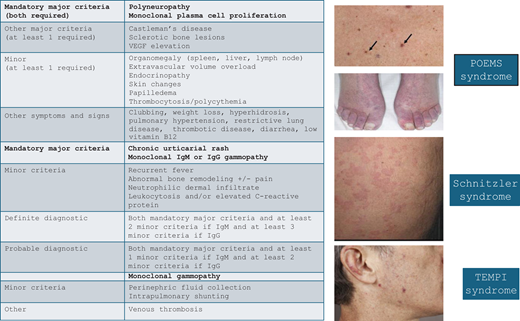
It cannot be overemphasized that in evaluating patients with monoclonal gammopathies, the diagnostic evaluation should include assessment of possible disorders beyond multiple myeloma, Waldenström macroglobulinemia, and light chain amyloidosis. While these entities are certainly important considerations in the appropriate context, excluding these limited diagnoses does not imply the patient has MGUS. Instead, MGCS must be considered systematically in the differential diagnosis of a patient with unexplained end-organ dysfunction and monoclonal gammopathy. In many cases, this diagnostic evaluation involves broad diagnostic studies to exclude other contributors to the clinical syndrome. In several MGCS subtypes, however, there are robust diagnostic systems or pathognomonic findings that confirm the causal association (Figure 3).
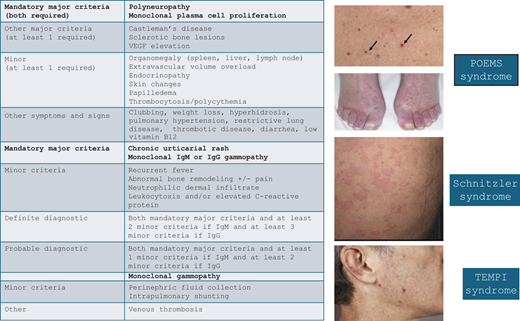
Diagnostic criteria and cutaneous manifestations of select multi-organ MGCS syndromes. MGCS, monoclonal gammopathy of clinical significance, VEGF, vascular endothelial growth factor. Adapted from: Dispenzieri A. How I treat POEMS syndrome. Blood. 2012;119(24):5650-5658, Figure 2; Palladini G, Merlini G. The elusive pathogenesis of Schnitzler syndrome. Blood. 2018;131(9):944-946, Figure; Sykes DB, O'Connell C, Schroyens W. The TEMPI syndrome. Blood. 2020;135(15):1199-1203, Figure 1.
Diagnostic criteria and cutaneous manifestations of select multi-organ MGCS syndromes. MGCS, monoclonal gammopathy of clinical significance, VEGF, vascular endothelial growth factor. Adapted from: Dispenzieri A. How I treat POEMS syndrome. Blood. 2012;119(24):5650-5658, Figure 2; Palladini G, Merlini G. The elusive pathogenesis of Schnitzler syndrome. Blood. 2018;131(9):944-946, Figure; Sykes DB, O'Connell C, Schroyens W. The TEMPI syndrome. Blood. 2020;135(15):1199-1203, Figure 1.
MGCS and multi-organ syndromes
While many types of MGCS are restricted to 1 organ system and are therefore somewhat easier to recognize, there are several complex MGCS syndromes with characteristic involvement of multiple organ systems and distinct pathophysiology. Here we describe 4 representative multi-organ MGCS syndromes.
Cryoglobulinemia
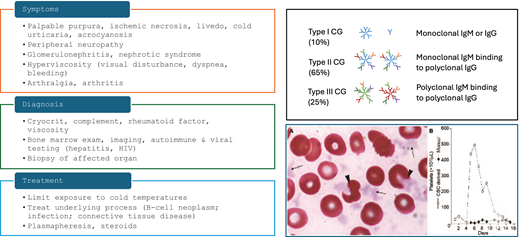
Several types of cryoglobulinemia (CG) exist, unified by the presence of cold precipitating immunoglobulins that form immune complexes and lead to vasculitis. Based on the Brouet classification, type 1 CG consists of monoclonal immunoglobulins, type 2 occurs when monoclonal immunoglobulins with rheumatoid factor activity complex with polyclonal IgG, and type 3 occurs in reactive states when polyclonal immunoglobulins lead to immune complex vasculitis.9 Type 1 and many type 2 CG patients have an underlying lymphoproliferative neoplasm, whereas rheumatologic disorders and infections such as hepatitis C are responsible for some type 2 and most cases of type 3 CG.
The presentation of CG is heterogeneous but commonly includes skin manifestations such as livedo and painful ulceration, as well as neuropathy and arthritis (Figure 4). Renal involvement may occur but is relatively uncommon.10
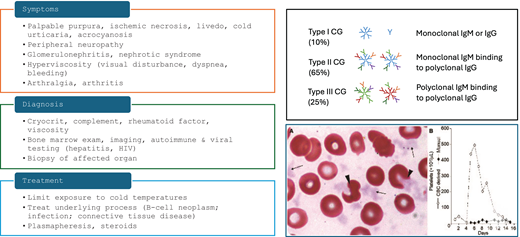
Diagnostic and therapeutic approach to cryoglobulinemia. The peripheral blood smear shows cryoglobulin precipitates mimicking platelets as well as altered erythrocyte morphology due to cryoglobulin attached to the red cell surface. Herishanu Y; Katz B-Z; ASH image bank 60686. CG, cryoglobulin.
Diagnostic and therapeutic approach to cryoglobulinemia. The peripheral blood smear shows cryoglobulin precipitates mimicking platelets as well as altered erythrocyte morphology due to cryoglobulin attached to the red cell surface. Herishanu Y; Katz B-Z; ASH image bank 60686. CG, cryoglobulin.
For treatment of patients with types 1 and 2 CG, identifying the lymphoproliferative clone responsible for the monoclonal gammopathy is paramount, as clone-targeting therapy is necessary to slow or eliminate the production of the toxic paraprotein. In many cases, the lymphoproliferative neoplasm is a lymphoplasmacytic clone producing IgM, although plasma-cell neoplasms may also occur.
The patient in our case had type 1 CG with painful cutaneous ulcers. Immunofixation of the cryoprotein demonstrated a pathogenic monoclonal IgM kappa immunoglobulin, with an underlying kappa-restricted plasma-cell neoplasm. Empiric steroids only temporarily relieved symptoms, and durable relief was seen with modest-intensity therapy directed against the plasma-cell clone using bortezomib and later daratumumab. Had she received only empiric therapy without identifying the underlying clonal disorder—for example, with rituximab targeting a putative B-cell clone responsible for the IgM—she likely would have experienced little relief, if any. Instead, she has enjoyed several years of excellent quality of life with low-intensity, time-limited therapy.
Schnitzler syndrome
The French dermatologist Dr. Liliane Schnitzler first recognized the rare acquired autoinflammatory syndrome that now carries her name. Schnitzler syndrome is a systemic disorder marked by constitutional symptoms, urticaria, sclerotic bone lesions, and monoclonal gammopathy, nearly always of the IgM kappa subtype. The disease usually presents in the fifth decade of life and affects males slightly more often than females. Additional symptoms include enlargement of the liver, spleen, and/or lymph nodes. The characteristic urticarial rash is intermittent and frequently changes distribution. There is no pathognomonic test to demonstrate causality between the paraprotein and the disease; consequently, the diagnosis depends on exclusion of other causes and the use of clinicopathologic criteria, which are known as the Strasbourg criteria.11
The disease is characterized by activation of the inflammasome and appears to be driven by IL-1, the levels of which are uniformly elevated. It is unclear whether the monoclonal protein triggers the autoinflammatory state or results from specific immune activation pathways. While many subtypes of MGCS are successfully treated with therapies targeting the underlying clonal population, the treatment of choice for Schnitzler syndrome is IL-1 blockade. The anti-IL-1 antibody anakinra rapidly improves disease symptoms, often within hours, with modest toxicity.12 Novel anti-IL-1 antibodies such as canakinumab have also been shown to work.13 IL-1 blockade is so effective that the lack of response in a patient with suspected Schnitzler syndrome should lead to reconsidering the diagnosis.
POEMS syndrome
Like Schnitzler syndrome, POEMS is a rare paraneoplastic syndrome mediated by cytokine secretion, namely vascular endothelial growth factor (VEGF). The acronym refers to the characteristic link between polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes. Patients with POEMS usually present in their 50s, and there is a slight male predominance. The hallmark of the disease is a symmetric ascending peripheral sensorimotor polyneuropathy. In addition to the signs and symptoms captured by the POEMS acronym, patients can also develop papilledema, extravascular volume overload, sclerotic bone lesions, and thrombocytosis/ erythrocytosis (PEST).14
The monoclonal immunoglobulin found in POEMS is almost exclusively of the lambda subtype. Another unique finding is the marked increase of serum VEGF secretion that plays a critical role in the disease pathogenesis. Abnormally high VEGF levels cause increased vascular permeability resulting in edema, including swelling of the myelin sheath, and increased angiogenesis may be responsible for the development of cutaneous angiomata. The connection between the lambda subtype of the clonal immunoglobulin and VEGF production is not fully understood but may involve an autoantibody mechanism affecting VEGF production.15
Osteosclerotic bone lesions are usually present and often focal. In some cases radiation of focal bone lesions can result in efficient disease control and resolution of symptoms. Systemic therapy has traditionally often included autologous stem cell transplant, but other plasma-cell-directed therapies, such as lenalidomide/dexamethasone, or more recently, proteasome-inhibitors and CD38-targeting antibodies, have also been used successfully.16
TEMPI syndrome
The first patient with TEMPI syndrome was described in 201017 followed by several case series. The characteristic manifestations of the disease are captured in the pentad of telangiectasia, elevated erythropoietin and erythrocytosis, monoclonal gammopathy, perinephric fluid collections, and intrapulmonary shunting. Patients most commonly present with telangiectasias of the face, torso, and upper extremities and almost always have very high erythropoietin levels.
Due in part to the rarity of the syndrome, the pathogenesis of TEMPI is not well elucidated. Unlike POEMS, the type of the monoclonal gammopathy does not favor lambda nor kappa. The possibility of renal damage by the monoclonal protein resulting in hypoxia and elevated erythropoietin levels is being considered. The erythropoietin level increases over time, usually before the development of perinephric fluid collections and intrapulmonary shunting, which in turn causes severe hypoxemia.
Therapy targeting the underlying plasma-cell clone can reverse the clinical syndrome. Successful use of bortezomib, lenalidomide, daratumumab, and autologous stem cell transplant has been reported, further supporting a causal relationship between the monoclonal gammopathy and TEMPI syndrome.18
Hematologic MGCS
A growing number of MGCS syndromes have been recognized that affect the hematologic system. These include acquired von Willebrand syndrome, cold agglutinin disease (CAD), and monoclonal gammopathy of thrombotic significance (MGTS).
Cold agglutinin disease
CAD most commonly occurs in older adults with an associated IgM kappa monoclonal gammopathy and an underlying B-cell or lymphoplasmacytic neoplasm. The MYD88 L265P variant may occur, indicating some overlap with Waldenström macroglobulinemia. Pentameric IgM binds the I antigen on red cells at temperatures in the thermal range of the autoantibody, which fixes complement and leads to largely extravascular hemolysis in hepatic Kupffer cells. The diagnosis is based on red cell agglutination, laboratory evidence of hemolytic anemia, and demonstration of a high-titer cold agglutinin, generally ≥1:64 with a clinically significant thermal amplitude ≥30 °C.19
Therapy has for years hinged on limiting production of the pathogenic paraprotein. Rituximab monotherapy is frequently effective as a first-line therapy for a variety of CD20+ lymphoproliferative disorders. For patients with refractory disease, more targeted therapies may be considered, such as bendamustine/ rituximab for those with lymphoplasmacytic lymphoma, or Bruton tyrosine kinase inhibitor or venetoclax-based combinations for those with underlying chronic lymphocytic leukemia. Steroids may temporize but rarely yield long-term disease control. Splenectomy is ineffective and therefore contraindicated.
In recent years, complement inhibition has emerged as an important adjunctive therapy in patients with CAD. The complement inhibitor sutimlimab blocks complement factor C1s and the ensuing complement-mediated hemolysis responsible for CAD.20 Sutimlimab can be given concurrently with clone-directed therapy, and a subset of patients are able to discontinue complement inhibition.
Monoclonal gammopathy of thrombotic significance
MGTS is a recently identified thrombotic disorder that shares many features of heparin-induced thrombotic thrombocytopenia (HITT) but does not require heparin exposure.21 Instead, autoantibody activity against the platelet factor 4 (PF4) antigen on platelets is mediated through a monoclonal immunoglobulin, typically IgG, which leads to platelet activation and clot formation. Published cases are few but share a common presentation of recurrent, often severe thrombosis in atypical sites, and without identifiable risk factors. Conventional heparin-induced thrombotic thrombocytopenia diagnostics with ELISA-based PF4 antibody testing and serotonin release assays are positive; a causal association with the monoclonal gammopathy is observed when the isolated anti-PF4 antibodies are demonstrated to share the same molecular mass as the serum monoclonal protein.
Therapy for MGTS includes anticoagulation as well as antineoplastic therapy that targets the underlying clonal proliferation. Plasma exchange and the off-label use of antibody-depleting therapies such as the neonatal Fc receptor inhibitor efgartigimod22 and the IgG-degrading enzyme imlifidase may also be considered.
Conclusions
MGCS defines a heterogeneous group of disorders in which disease manifestations are governed by a toxic monoclonal paraprotein produced by an indolent lymphoproliferative clone. Common syndromes principally affect renal, neurologic, and cutaneous organ systems, although hematologic and other systemic syndromes are also recognized. Understanding the workup of monoclonal gammopathy, its epidemiology, and assays to evaluate the causality between the monoclonal protein and the clinical syndrome is critical to the clinicopathologic diagnosis of these disorders. Therapy often relies on clone-directed therapy, although occasionally patients may benefit from cytokine blockade or antibody-depleting therapies.
Conflict-of-interest disclosure
David Iberri reports no competing financial interests to declare.
Michaela Liedtke reports research funding from Caelum, Janssen, BMS, Gilead, AbbVie, Allogene, and Seagen and Biomea; has participated in advisory committees for AbbVie, Kite, and BMS; and is on the scientific advisory board for Nexcella.
Off-label drug use
David Iberri: The authors discuss off-label drug use.
Michaela Liedtke: The authors discuss off-label drug use.