
Abstract
Beta-thalassemia is a genetic disorder with mutations in the β-globin gene that reduce or abolish β-globin protein production. Patients with β-thalassemia major (Cooley's anemia) become severely anemic by 6 to 18 months of age, and are transfusion dependent for life, while those with thalassemia intermedia, a less-severe form of thalassemia, are intermittently or rarely transfused. An allogeneically matched bone marrow transplant is curative, although it is restricted to those with matched donors. Gene therapy holds the promise of “fixing” one's own bone marrow cells by transferring the normal β-globin or γ-globin gene into hematopoietic stem cells (HSCs) to permanently produce normal red blood cells. Requirements for effective gene transfer for the treatment of β-thalassemia are regulated, erythroid-specific, consistent, and high-level β-globin or γ-globin expression. Gamma retroviral vectors have had great success with immune-deficiency disorders, but due to vector-associated limitations, they have limited utility in hemoglobinopathies. Lentivirus vectors, on the other hand, have now been shown in several studies to correct mouse and animal models of thalassemia. The immediate challenges of the field as it moves toward clinical trials are to optimize gene transfer and engraftment of a high proportion of genetically modified HSCs and to minimize the adverse consequences that can result from random integration of vectors into the genome by improving current vector design or developing novel vectors. This article discusses the current state of the art in gene therapy for β-thalassemia and some of the challenges it faces in human trials.
Rationale for Genetic Therapy
Beta thalassemias are among the most common single-gene defects worldwide, and pose a severe health and economic burden to patients and families at risk. They are caused by different mutations in the β-globin gene cluster, resulting in reduced or absent adult hemoglobin (HbA), which is composed of two alpha- and two beta-globin chains (α2β2), leading to severe anemia. This disorder is characterized by reduced HbA production (β+ thalassemia) or complete absence of β-globin synthesis (β0 thalassemia). In humans, the degree of imbalance in the α- and β-globin protein is directly linked to the clinical severity of β-thalassemia. This imbalance also hinders erythroid precursor maturation, resulting in ineffective erythropoiesis. Humans that are heterozygotic for β-thalassemia mutations are very mildly anemic and lead normal lives. Homozygosity results in disease: thalassemia intermedia, in which individuals are moderately anemic due to reduced β-globin production, and thalassemia major or Cooley's anemia, characterized by absent or severely reduced β-globin production and complete transfusion dependence for survival. Patients with severe β-thalassemia major show significant levels of iron accumulation in multiple organs and tissues from transfusional hemosiderosis, and inadequately transfused patients develop massive erythroid hyperplasia and extramedullary hematopoiesis in the body's attempt to compensate for the red blood cell loss. Iron overload, associated with chronic transfusions, is the major cause of death.1
Hematopoietic stem-cell (HSC) transplantation using bone marrow, umbilical cord blood, or mobilized peripheral blood as a source of HSCs has been performed in numerous patients with thalassemia. The results of HSC transplant are remarkable if a matched sibling donor is available and the transplants are performed in well-transfused and chelated children with good organ status. The accompanying review by Dr. Angelucci summarizes the current status of transplantation for thalassemia (see p. 456 in this issue).
HSC transplant is limited by the availability of matched donors and is associated with immunological complications, including graft rejection and graft-versus-host disease.2–4 Gene transfer using autologous bone marrow can potentially permanently cure thalassemia major, and could resolve the limitations of finding a matched donor and eliminate the risks of graft-versus-host disease and graft rejection that are associated with allogeneic bone marrow transplantation.
Elements Required for High Level of β-Globin Expression
For gene transfer of a normal β-globin gene to be an option, several conditions must be met, including: 1) efficient gene transfer and engraftment of a high number of HSCs, 2) regulated expression in the erythroid lineage, 3) consistent and therapeutic levels of β-globin gene expression, and 4) safe levels of expression, ideally devoid of position effects.
Earlier studies on globin gene regulation and gamma-retroviral globin gene-encoding vectors were pivotal in providing critical insights into elements required for the high-level, regulated expression of globin genes. Gamma-retroviral vectors were limited by their size, stable transmission, and high level of expression of the β-globin gene due to: 1) the requirement for large elements of the locus control region (LCR) for high level expression; initial efforts to incorporate minimal core LCR elements resulted in vectors with low titers5 and low levels of expression that were prone to position effects6 ; 2) transcriptional interference of the retroviral long terminal repeats (LTR) with LCR elements resulted in unstable proviral transmission with vectors prone to sequence rearrangements7 and poor transgene expression.8–9 ; and 3) the β-globin gene is an intron-dependent gene and needs to be inserted in reverse orientation in viral vectors.10–11 Several modifications allowed efficacious expression: 1) deletion of deleterious sequences in intron 2 of the β-globin gene to allow stable transmission and expression of β-globin gene from retroviral vectors (Figure 1A)12–13 ; 2) the β-globin cDNA was used with post-transcriptional regulatory elements14 ; 3) the α-locus HS-40 regulatory region was used in place of the large β-globin LCR15–18 ; and 4) alternative erythroid-specific promoters such as ankyrin19–20 and mutant hereditary persistence of fetal hemoglobin (HPFH) γ-globin promoters were used,21–22 but had limited success. Studies by Emery et al.23 and Rivella et al.24 showed that incorporation of the chicken hypersensitive site-4 insulator (cHS4) element in gamma-retrovirus vectors improved the performance of the β-globin transgene expression to a significant extent, although its therapeutic relevance was not studied.

(A) Prototypic design of a γ-retroviral vector β/γ globin gene and the small LCR core elements. The β-globin gene (exons depicted in blue shaded boxes) is placed in the reverse orientation in the retroviral vector to prevent splicing of the introns in the viral genomic RNA during production of the virus. Arrow indicates the direction of transcription. In the γ-retroviral vector construct, HS2, HS3, and HS4 denotes LCR core derivatives.12 (B) Successful β/γ-globin LV vectors used by different groups. TNS9 vector is based on the publication by May et al.24 and encodes the human beta globin gene. The βA87Q vector encodes a mutated β-globin gene at the 87th codon, conferring it with anti-sickling properties,55 and also carries two copies of the 5′ 250-bp cHS4 insulator core. This vector was used for the gene therapy trial for β-thalassemia in France led by Dr. Philippe Leboulch. The BG-I vector, described by Putheenvetil et al.,32 expresses β-globin from a 254-bp β-globin promoter, contains large LCR elements, and is flanked by the 1.2-kb full-length cHS4 insulator in the LTRs. The vector d432β-Aγ vector28 also contains γ-globin 3′ regulatory untranslated regions (shaded purple) and enhancer elements downstream of the γ-globin coding sequences (3′e*). mLARβΔγV5 vector30 consists of extended regions of β-globin LCR. The GLOBE vector is described in the article by Miccio et al.29 Triangles represent deletion of the 372-bp purine-rich sequences in the β-globin IVS 2 (a 562-bp deletion of IVS2 in the GLOBE vector) and the 400-bp enhancer deletion in the LTR to generate a self-inactivating vector design. RRE, rev response element; HS2, HS3, and HS4, DNase hypersensitive sites 2, 3, and 4 present in the β-globin LCR and the length of respective hypersensitive sites are indicated adjacent to them; cPPT, central polypurine tract; LTR, long terminal repeat; 3′e, β-globin 3′ enhancer element; ψ, packaging signal; Ψ+, extended packaging signal; βP, β-globin gene promoter. The β or γ globin genes are depicted in the reverse orientation, with β-globin exons in blue, γ-globin exons in green, and β-globin introns shown as unshaded boxes
(A) Prototypic design of a γ-retroviral vector β/γ globin gene and the small LCR core elements. The β-globin gene (exons depicted in blue shaded boxes) is placed in the reverse orientation in the retroviral vector to prevent splicing of the introns in the viral genomic RNA during production of the virus. Arrow indicates the direction of transcription. In the γ-retroviral vector construct, HS2, HS3, and HS4 denotes LCR core derivatives.12 (B) Successful β/γ-globin LV vectors used by different groups. TNS9 vector is based on the publication by May et al.24 and encodes the human beta globin gene. The βA87Q vector encodes a mutated β-globin gene at the 87th codon, conferring it with anti-sickling properties,55 and also carries two copies of the 5′ 250-bp cHS4 insulator core. This vector was used for the gene therapy trial for β-thalassemia in France led by Dr. Philippe Leboulch. The BG-I vector, described by Putheenvetil et al.,32 expresses β-globin from a 254-bp β-globin promoter, contains large LCR elements, and is flanked by the 1.2-kb full-length cHS4 insulator in the LTRs. The vector d432β-Aγ vector28 also contains γ-globin 3′ regulatory untranslated regions (shaded purple) and enhancer elements downstream of the γ-globin coding sequences (3′e*). mLARβΔγV5 vector30 consists of extended regions of β-globin LCR. The GLOBE vector is described in the article by Miccio et al.29 Triangles represent deletion of the 372-bp purine-rich sequences in the β-globin IVS 2 (a 562-bp deletion of IVS2 in the GLOBE vector) and the 400-bp enhancer deletion in the LTR to generate a self-inactivating vector design. RRE, rev response element; HS2, HS3, and HS4, DNase hypersensitive sites 2, 3, and 4 present in the β-globin LCR and the length of respective hypersensitive sites are indicated adjacent to them; cPPT, central polypurine tract; LTR, long terminal repeat; 3′e, β-globin 3′ enhancer element; ψ, packaging signal; Ψ+, extended packaging signal; βP, β-globin gene promoter. The β or γ globin genes are depicted in the reverse orientation, with β-globin exons in blue, γ-globin exons in green, and β-globin introns shown as unshaded boxes
Genetic Therapy for β-Thalassemia with Lentiviral Vectors
In the mid 1990s, lentiviral (LV) vectors derived from HIV made possible the development of globin LV vectors. LV vectors can package full-length, unspliced RNA due to the presence of a strong RNA export element, the rev response element (RRE), and can carry large transgene cassettes with introns and regulatory LCR elements. Moreover, globin LV vectors can stably transmit globin regulatory elements and the coding sequences of the β-globin gene. The promise of “additive gene therapy” as a therapeutic option for β-thalassemia was therefore made possible with these vectors for the first time in the field of gene transfer for the treatment of hemoglobinopathies.
Correction of Murine Thalassemia
In a pioneering study, May et al. showed phenotypic correction of thalassemia mice (Hbbth3/+ mice that resemble human thalassemia intermedia phenotype), with an average increase of hemoglobin by 3 to 4 g/dL per vector copy with the TNS9 vector. Subsequently, others have shown correction of murine thalassemia using similar β- or γ-globin-encoding LV vectors (Figure 1B and Table 1).25–29 A major finding that emerged from these studies was that thalassemia mice with an average copy number of one did not show correction, and multiple copies were required for a therapeutic effect.27 Variable globin transgene expression was evident in thalassemia mice,26,28 which was often sub-therapeutic for β0-thalassemia.26 Therefore, subsequent studies focused on higher and more consistent expression for effective correction of thalassemia major.
Comparisons of different β- or γ-globin vectors studied successfully in mouse and human models of β-thalassemia
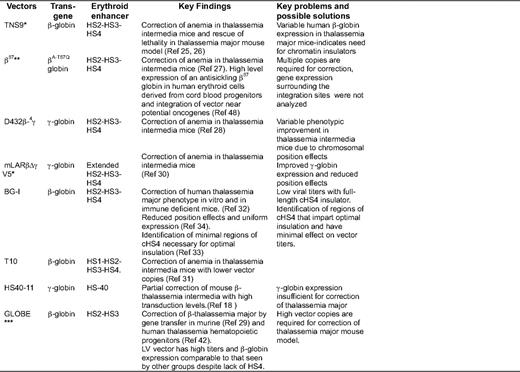
Status of clinical trials denoted by asterisks: *US trial planned using this vector or with minor modifications; **clinical trial ongoing in France; ***clinical trial planned in Europe
Improved Expression and Position Effects of LV Vectors
Higher and more consistent globin expression at a lower copy number was addressed by different groups using several strategies. Hanawa et al. included larger HS2–4 elements of the LCR.30 Lisowski et al.31 showed that inclusion of HS1, in addition to HS2–4, improved the overall expression. Puthenveetil et al.32 and Arumugam et al.33–34 from our group, and Emery et al.35–36 flanked the LV and γ-retrovirus vectors, respectively, with a chromatin insulator from cHS4. Chromatin insulators are DNA elements that can shield the genes from their surrounding chromatin environment. Insulators exert two functions: they prevent gene silencing from the effect of heterochromatin (barrier activity), and they prevent activation of a gene promoter by an adjacent enhancer (enhancer blocking activity).37 Incorporation of insulator elements in vectors can therefore function to diminish position effects and silencing and block surrounding gene enhancers.38 We showed correction of β0 thalassemia major (Cooley's anemia) in the human erythroid progeny of human CD34+ cells in vitro and in xenografts, and in the red blood cell progeny of thalassemia intermedia murine HSC using cHS4-insulated LV vector BG-I.33–34
Although insulated vectors provided consistent/uniform expression, and therefore lead to superior correction, the caveat is that LV vectors with the full-length (1.2 kb) cHS4 element in the LTR exhibit marked reductions in infectious titers.39 LV vectors carrying the large globin gene and LCR have modest titers even without insertion of large insulator elements in the LTR. The further sharp decline in titers limits the large-scale production of vector for human trials. The smaller core elements of cHS4 were therefore explored, but the core does not retain the insulator activity of the full-length insulator.33,36 The cHS4 insulator can provide consistent expression of globin genes33–35 and can impart enhancer-blocking effects40–41 if its effect on viral titers can be overcome.39 Miccio et al.29 generated LV vectors with high titers by shortening the globin insert in the vector. They removed HS4, retaining only HS2 and HS3 of the LCR, and removed a larger portion of intron 2 of the β-globin gene. This vector has high titers and is able to efficiently correct murine thalassemia (and more recently, human thalassemia intermedia and major42 ), although high vector copies are required to correct murine thalassemia major with this vector (Table 1)
Chimerism of Genetically Modified HSCs Required for Correction of Cooley's Anemia
Most murine transplant studies are performed with a myeloablative regimen that allows near-complete donor engraftment, and thus there is high chimerism of genetically modified HSCs. Clinically, unless myeloablative conditioning is used, this may not be necessary. Persons et al. showed that chimerism levels of only 10% to 30% normal HSCs are sufficient to result in nearly complete hematologic and pathologic correction of thalassemia mice,43 and our group has shown that 15% to 20% genetically corrected HSCs are sufficient to correct sickle-cell disease.44 This level of chimerism require a significant amount of pre-transplant chemotherapy conditioning for cytoreduction of endogenous HSCs, a large stem cell dose, and a high proportion of genetically modified HSCs. This is in contrast to gene-transfer approaches for immunodeficiency disorders, in which minimal to no cytoreduction is sufficient for phenotypic correction due to the tremendous selective advantage of gene-modified T-cell precursors and T cells.
Gene Therapy Clinical Trials in Thalassemia
The status of potential clinical trials in the upcoming future and an open clinical trial for β-thalassemia is included in Table 1. Leboulch et al. have opened a trial in France that has enrolled two patients. One patient with E-β0-thalassemia was transplanted with CD34+ cells transduced with the β-globin vector (Figure 1B) following myeloablative pre-transplant conditioning, and has shown sustained improvement over 2 years in hemoglobin and transfusion independence. At a molecular level, this has occurred due to selective proliferation of one myelo-erythroid clone with a viral integration that has apparently conferred this clone a proliferative advantage, as described in the subsequent section.
Genotoxicity with Integrating Viral Vectors
A safety concern is the risk of insertional mutagenesis with randomly integrating viral vectors. Insertional oncogenesis by the viral LTR enhancer has been shown in humans, in the context of gene therapy for X-linked severe combined immunodeficiency syndrome (X-SCID1) with γ-retroviral vectors in four patients in the French X-SCID1 trial45 and one patient in the London X-SCID1 trial,46 despite a very high degree of success with gene transfer. Four of five patients showed insertional activation of the LMO2 oncogene. Recently, in a similarly successful chronic granulomatous disease trial in Germany, insertional activation of MDS-EVI-1 by the viral LTR enhancer resulted in myelodysplastic syndrome in two patients that was accompanied by the silencing of the transgene expression due to methylation of CpG island in the viral LTR promoter, resulting in the abolishment of a therapeutic effect of the transgene.47 A study by Imren et al.48 with β-globin LV vector-marked human hematopoietic cells in immune-deficient mice showed that 86% of insertions occurred within genes, including genes involved in hematopoiesis and in human leukemia, although genetic dysregulation of these genes was not addressed in this study. Hargrove et al. showed direct evidence that globin LV vectors can cause insertional dysregulation of cellular genes in primary, clonal, murine, β-thalassemic erythroid cells in which the globin locus control region was active.49 Similar results were published subsequently by Arumugam et al. using the in vitro immortalization assay in primary murine hematopoietic progenitors.50 The French trial described below has now confirmed the LCR-mediated cellular gene activation in a patient with hemoglobin E-β0 thalassemia.49
In the first trial for gene transfer for the treatment of β-thalassemia, Leboulch et al. observed a therapeutic effect in one patient that was associated with the emergence of a dominant erythroid clone with insertion of the vector in the HMGA2 gene locus.51 The patient had approximately 10% gene marked hematopoiesis, of which a large percentage is contributed by this clone. The HMGA2 gene expression is increased in erythroid cells and not in myeloid cells, indicating dysregulation of the gene by the LCR in erythroid lineage. At the burst-forming unit-erythroid (BFUe) level, the amount of β87 globin produced by this clone is similar compared with other gene-marked BFUe. So far, this expansion of the dominant clone has resulted in a therapeutic benefit for 2 years following gene transfer, and there has been no perturbation of hematopoiesis. This LV had two copies if the cHS4 “core” that recombined and the integrated LV is flanked by one copy of the recombined cHS4 core (reported by Leboulch at the American Society of Gene Therapy Meeting, 2009; at the 5th Stem Cell Clonality and Genotoxicity Retreat, 2009; and at the Recombinant DNA Advisory Committee; for the latter, a webcast is available online at: http://oba.od.nih.gov/rdna/rac_past_meeting_2009_webcasts.html#dec09).51 At this point, the cause and effect are unknown and the field awaits further developments.
Safety of Integrating LV for β-Thalassemia
Using a sensitive in vitro immortalization assay, we have explored the genotoxicity of the β-globin LCR enhancer and promoter, with and without different sized cHS4 insulator elements in the LTR of the self-inactivating LV using primary hematopoietic progenitors to generate in vitro immortalization assay mutants. LCR carrying LV had approximately 200-fold lower transforming potential compared with the conventional γ-retroviral vectors. A further 3-fold reduction in transforming activity was observed with the LCR containing LV carrying either the full-length cHS4 or the 650-bp cHS4 (comprising of the 5′ 250 bp core and the distal3′ 400 bp).50 Our data indicate that toxicology studies of LCR containing LV in mice will likely not yield insertional oncogenesis with the number of mice that can be practically studied.
The clonal dominance seen in the patient with β-thalassemia treated in France suggests that the cHS4 core alone is unable to shield the HMGA2 gene from the LCR enhancer. Recently, a structure-function analysis of cHS4 showed unique insulator activity in the distal 400-bp cHS4 sequences, which, when combined with the 5′ core, restored full barrier31 and enhancer blocking46 activity. Overall, the identification of more potent insulators is necessary to impart safety to randomly integrating vectors.
Alternate Genetic Approaches for Correcting β-Thalassemia
Most traditional gene-therapy strategies add the function of a full-length gene to a population of target cells while the endogenous mutant gene is still present. Genetic correction using homologous recombination of the β-sickle globin has been done in mouse embryonic stem cells and induced pluripotent stem cells reprogrammed from fibroblasts in a sickle cell anemia mouse model. The corrected induced pluripotent stem cells were then differentiated into HSCs and transplanted, with complete correction of sickle-cell anemia.52–53 The reprogramming of somatic cells into induced pluripotent stem cells opens a new approach for treating β-thalassemia.54 Ye et al. reprogrammed the skin fibroblasts of a patient with homozygous β° thalassemia into induced pluripotent stem cells, and showed that these cells could be differentiated into hematopoietic cells that synthesized hemoglobin. These reports suggest that induced pluripotent stem cells could offer a new approach for the treatment of β-thalassemia. These approaches are at an experimental level at the present time.
Summary
The therapeutic efficacy of LV-mediated globin gene transfer in HSC has been adequately demonstrated over the past decade, resulting in the correction of mouse and human models of Cooley's anemia and thalassemia intermedia. The safety of gene transfer has been the primary goal of the researchers in designing globin vectors. The design of lineage and differentiation stage-restricted vectors represents one major step in reducing the risk of trans-activating oncogenes. The risk of insertional oncogenesis can likely be reduced significantly by rational vector design with tightly regulating expression of the vector-encoded transgene and by minimizing interactions between vector elements and flanking transgenes with optimal insulator elements. In this respect, HSC gene transfer to treat β-thalassemia is now becoming a therapeutic reality. However, caution regarding the insertional mutagenesis capability of any randomly integrating vector remains, until homologous recombination and induced pluripotent stem cell technology becomes translatable to the clinic.
Acknowledgments
This work was supported by NIH grant no. U54 HL06–008.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Off-label drug use: None disclosed.
Correspondence
Punam Malik, MD, TCHRF 6564, Division of Experimental Hematology, Cincinnati Children's Hospital Medical Center, ML 7013, 3333 Burnet Avenue, Cincinnati, OH 45229; Phone: 513-636-0364; Fax: 513-636-3768; e-mail: Punam.Malik@cchmc.org
