
Abstract
The diagnosis of autoimmune hemolytic anemia (AIHA) can be made with a stepwise approach that aims to identify laboratory and clinical evidence of hemolysis and then determine the immune nature of hemolysis with the direct anti-globulin test. Once alternative causes for these findings have been excluded, AIHA is established, and the clinician must search for secondary causes, as well as identify the type of AIHA. Rituximab is now the preferred second-line treatment for primary warm AIHA and first-line treatment for primary cold agglutinin disease (CAD), either as monotherapy or combined with bendamustine. Complement inhibitors have shown utility in stabilizing AIHA patients with acute severe hemolysis. Future prospects are discussed and include the C1s inhibitor BIVV009 (sutimlimab) that is now entering phase 3 studies for CAD.
Learning Objectives
Understand how AIHA and its subtypes can be diagnosed through a combination of clinical assessment and laboratory tests
Describe the latest advances in treatment of primary warm AIHA and cold agglutinin disease
Approach to the diagnosis of autoimmune hemolytic anemia
Autoimmune hemolytic anemia (AIHA) is a decompensated acquired hemolysis caused by the host’s immune system acting against its own red cell antigens. Consequent complement activation can impact the clinical picture and is an emerging target for therapeutic approaches.
When a patient presents with anemia, a stepwise approach should be followed. Initial simple investigations (Table 1) will first alert the physician to the suggestion of hemolysis as the cause of the anemia. These include a normo-/macrocytic anemia, raised reticulocyte count, raised unconjugated bilirubin, reduced haptoglobin, and blood smear with polychromasia or more specific features, such as spherocytes or agglutination (Figure 1). Although the typical pattern is presented, none of these tests are fully sensitive or specific for hemolysis. For example, liver disease can increase lactate dehydrogenase (LDH) and reduce haptoglobin. The bilirubin may be normal with milder hemolysis, and spherocytes are not always visible. Reticulocytopenia can occur in AIHA, secondary to bone marrow infiltration by a lymphoproliferative disorder or to parvovirus B19 infection. However, reticulocytopenia is also observed in a significant minority of patients at presentation (20% in 1 series1 ), despite erythroid hyperplasia in the marrow. This may be caused by immune attack on late-stage erythroid precursors or reflect a lag in marrow responsiveness, but it can sometimes persist and predict a more severe clinical course.2 A significantly raised LDH, red cell fragments on smear, or the presence of urinary hemosiderin suggests a predominant intravascular component to the hemolytic process. There are fewer causes of intravascular hemolysis; therefore, these can be very useful for subsequent investigations (Table 2). Once hemolysis is confirmed, further investigation is needed to establish whether that hemolysis is immune, principally by the direct anti-globulin test (DAT). The standard DAT demonstrates that immunoglobulin G (IgG) and/or complement (usually C3d) is bound to the red cell membrane. Autoantibodies can also be of IgM and IgA classes, and, in some circumstances, an extended DAT panel can be used to detect these.
Investigations in patients presenting with AIHA
| Diagnostic tests | Indication for tests |
|---|---|
| Primary evaluation | |
| Hemolytic screen | |
| FBC, blood smear, LDH, haptoglobin, bilirubin, DAT, reticulocyte count with or without urine for hemosiderin or urine dipstick for microscopy | |
| Detection of underlying disorders (investigation of AIHA) | |
| Serum Igs and electrophoresis with immunofixation* | |
| HIV, HBV, HCV | |
| Anti-dsDNA, ANA | |
| CT chest, abdomen, and pelvis | |
| Additional investigation in selected patients with AIHA | |
| Bone marrow examination | CAD, age ≥ 60 years, features in history, examination, FBC or smear suggesting possible marrow infiltration |
| U&E, LFT, clotting, BP, urine dipstick | If pregnant or thrombocytopenic, to exclude DIC or pregnancy-associated TMA |
| Infection screening | Dependent on symptoms, travel history, and age |
| Peripheral T-cell subsets, creatinine, LFT, clotting | All children and if suspected Evans syndrome |
| Parvovirus, hematinics | If reticulocytopenia |
| Additional serological investigation in selected patients with AIHA | |
| DAggT | If DAT positive for C3d ± IgG |
| Cold antibody titer | If DAggT positive |
| Monospecific DAT for IgM, G, A, C3d | If DAT-negative AIHA suspected |
| Red cell eluate | If (monospecific) DAT-negative AIHA suspected |
| Donath Landsteiner | If DAT is positive for C3d ± IgG and |
| i) DAggT-negative or insignificant CAs and | |
| ii) Age < 18 y or hemoglobinuria or cold-associated symptoms or atypical serology | |
| Thermal range of cold antibody | If clinical significance of cold autoagglutinin unclear |
| Diagnostic tests | Indication for tests |
|---|---|
| Primary evaluation | |
| Hemolytic screen | |
| FBC, blood smear, LDH, haptoglobin, bilirubin, DAT, reticulocyte count with or without urine for hemosiderin or urine dipstick for microscopy | |
| Detection of underlying disorders (investigation of AIHA) | |
| Serum Igs and electrophoresis with immunofixation* | |
| HIV, HBV, HCV | |
| Anti-dsDNA, ANA | |
| CT chest, abdomen, and pelvis | |
| Additional investigation in selected patients with AIHA | |
| Bone marrow examination | CAD, age ≥ 60 years, features in history, examination, FBC or smear suggesting possible marrow infiltration |
| U&E, LFT, clotting, BP, urine dipstick | If pregnant or thrombocytopenic, to exclude DIC or pregnancy-associated TMA |
| Infection screening | Dependent on symptoms, travel history, and age |
| Peripheral T-cell subsets, creatinine, LFT, clotting | All children and if suspected Evans syndrome |
| Parvovirus, hematinics | If reticulocytopenia |
| Additional serological investigation in selected patients with AIHA | |
| DAggT | If DAT positive for C3d ± IgG |
| Cold antibody titer | If DAggT positive |
| Monospecific DAT for IgM, G, A, C3d | If DAT-negative AIHA suspected |
| Red cell eluate | If (monospecific) DAT-negative AIHA suspected |
| Donath Landsteiner | If DAT is positive for C3d ± IgG and |
| i) DAggT-negative or insignificant CAs and | |
| ii) Age < 18 y or hemoglobinuria or cold-associated symptoms or atypical serology | |
| Thermal range of cold antibody | If clinical significance of cold autoagglutinin unclear |
ANA, antinuclear antibody; BP, blood pressure; CAD, cold agglutinin disease; CAs, cold agglutinins; CT, computerized tomography; DAggT, direct agglutination test; DIC, disseminated intravascular coagulation; dsDNA, double-stranded DNA; FBC, full blood count; HBV, hepatitis B virus; HCV, hepatitis C virus; LFT, liver function tests; TMA, thrombotic microangiopathy; U&E, urea and electrolytes.
If a cold autoantibody suspected, keep sample at 37°C until serum has been separated.
Modified from Hill et al, with permission.12
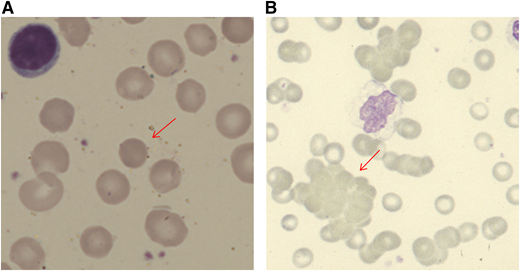
Blood film appearances in patients with AIHA (both using May Grunwald Giemsa stain). (A) Spherocyte in a patient with warm AIHA (original magnification, ×100). (B) Red cell agglutination in a patient with cold agglutinin disease (×40).
Blood film appearances in patients with AIHA (both using May Grunwald Giemsa stain). (A) Spherocyte in a patient with warm AIHA (original magnification, ×100). (B) Red cell agglutination in a patient with cold agglutinin disease (×40).
Causes of intravascular hemolysis
| Complement fixation (eg) |
| Paroxysmal nocturnal hemoglobinuria |
| Paroxysmal cold hemoglobinuria |
| Acute hemolytic transfusion reactions due to mismatched blood |
| Some autoimmune hemolytic anemias if complement fixation allows terminal complement activation (membrane attack complex formation) on the surface of red blood cells causing lysis |
| Mechanical trauma (eg) |
| Cardiac valves |
| Microthrombi in circulation - DIC, MAHA (TTP, HUS, aHUS) |
| March hemoglobinuria |
| Toxic or exogenous factors (eg) |
| Infections: Babesia, Clostridium, Leptospira, Falciparum malaria |
| Spider bites |
| Snake venoms |
| Copper or zinc toxicity |
| Hypotonic solutions |
| Complement fixation (eg) |
| Paroxysmal nocturnal hemoglobinuria |
| Paroxysmal cold hemoglobinuria |
| Acute hemolytic transfusion reactions due to mismatched blood |
| Some autoimmune hemolytic anemias if complement fixation allows terminal complement activation (membrane attack complex formation) on the surface of red blood cells causing lysis |
| Mechanical trauma (eg) |
| Cardiac valves |
| Microthrombi in circulation - DIC, MAHA (TTP, HUS, aHUS) |
| March hemoglobinuria |
| Toxic or exogenous factors (eg) |
| Infections: Babesia, Clostridium, Leptospira, Falciparum malaria |
| Spider bites |
| Snake venoms |
| Copper or zinc toxicity |
| Hypotonic solutions |
aHUS, atypical hemolytic uremic syndrome; DIC, disseminated intravascular coagulation; HUS, hemolytic uremic syndrome; MAHA, microangiopathic hemolytic anemia; TTP, thrombotic thrombocytopenic purpura.
After identifying hemolytic anemia with a positive DAT, hopefully obvious causes, such as a delayed transfusion reaction from a recent transfusion, alloimmune hemolysis following solid organ or allogeneic stem cell transplantation, drug-induced immune hemolysis, or hemolytic disease of the newborn, will be quickly identified by clinical assessment. When considering alternatives, it should be remembered that a positive DAT can occur as a result of other processes, such as passive deposition of antibodies or immune complexes in liver disease, chronic infection, malignancy, systemic lupus erythematosus (SLE), renal disorders, and following drug therapies, such as IV Ig or anti-thymocyte globulin. If no alternative cause is identified, a diagnosis of AIHA can be made. AIHA can occur infrequently with a negative DAT by standard techniques (eg, due to a low-affinity antibody or an IgA-only antibody). A diagnosis of DAT-negative AIHA can be made following careful exclusion of alternative causes of hemolysis, and confirmation by a sensitive technique where available, and is supported by a response to steroid therapy.
The next step is to investigate for an underlying associated condition, which is found in ∼50% of patients, along with further serological tests to determine the type of AIHA, because the approach to treatment differs (Table 1). A suggested diagnostic pathway for AIHA is shown in Figure 2, although it is recognized that there are exceptions (eg, a low-titer cold antibody can sometimes be clinically significant). Therefore, a final diagnosis may require synthesis of the clinical picture and advice from a specialist reference laboratory.
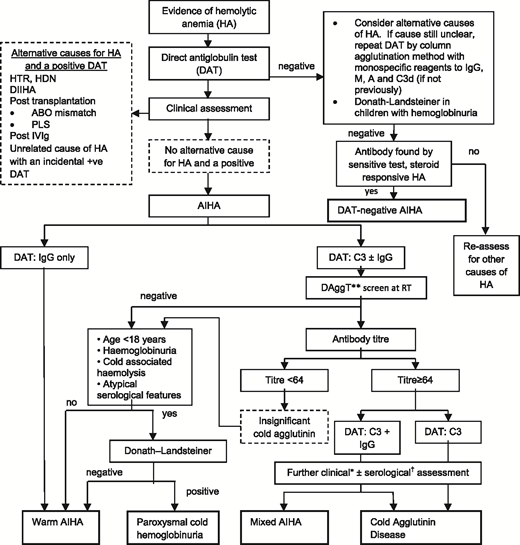
Diagnostic pathway for AIHA. DAggT, direct agglutination test; DIIHA, drug-induced immune hemolytic anemia; HA, hemolytic anemia; HDN, hemolytic disease of the newborn; HTR, hemolytic transfusion reaction; PLS, passenger lymphocyte syndrome; RT, room temperature. *The final diagnosis of CHAD or mixed AIHA is based on the overall clinical picture, including supportive serological findings. †For example, the thermal amplitude. **Saline-suspended red cells and patient’s serum at room temperature for 30 to 60 minutes. Adapted from Hill et al with permission.12
Diagnostic pathway for AIHA. DAggT, direct agglutination test; DIIHA, drug-induced immune hemolytic anemia; HA, hemolytic anemia; HDN, hemolytic disease of the newborn; HTR, hemolytic transfusion reaction; PLS, passenger lymphocyte syndrome; RT, room temperature. *The final diagnosis of CHAD or mixed AIHA is based on the overall clinical picture, including supportive serological findings. †For example, the thermal amplitude. **Saline-suspended red cells and patient’s serum at room temperature for 30 to 60 minutes. Adapted from Hill et al with permission.12
Approximately 65% of patients have a warm AIHA; this can be diagnosed in patients with a consistent clinical picture and a DAT positive for IgG only or when DAT is positive for C3d ± IgG, when a clinically significant cold antibody has been excluded.
Primary cold agglutinin disease (CAD) is caused by an underlying lymphoproliferative bone marrow disorder, and its pathophysiology is described elsewhere.3 Hemolysis is caused by IgM antibodies that are most active in vitro at low temperatures. In vivo, these antibodies bind red cells in the cooler peripheral circulation, causing agglutination that leads to acrocyanosis or Raynaud’s disease in some patients. IgM binds C1q on the red cell membrane, activating the classical complement pathway but disassociating from the red cells centrally, leading to a DAT that is characteristically positive for only C3d, although a weak positive IgG is sometimes also detected. This process leads to extravascular removal of C3b-coated red cells by the reticuloendothelial system; however, exacerbations can activate C5, leading to intravascular hemolysis. In patients with a DAT positive for C3d ± IgG, a direct agglutination test can be performed as a screening test in the local transfusion laboratory, and a clinically significant cold agglutinin can be excluded if saline-suspended normal red cells are not agglutinated by the patient’s serum after incubation at room temperature for 30 to 60 minutes. Patients with positive screening can then have antibody titers assessed. CAD can be diagnosed in patients with AIHA and a DAT positive for C3d ± IgG, with a consistent clinical picture and a high-titer cold reactive antibody (titer ≥ 1:64 at 4°C). Assessment of the antibodies’ thermal amplitude can be useful and is usually ≥30°C when red cells are suspended in 30% bovine albumin. Serum electrophoresis will usually detect a monoclonal paraprotein, typically IgMκ, but serum must be kept at 37 to 38°C from the time of sampling until serum is removed from the clot.
Cold agglutinin syndrome (CAS) can be diagnosed in patients with laboratory criteria consistent with a clinically significant cold antibody that occurs in association with secondary disorders, such as infection, SLE, or aggressive lymphoma.
Mixed AIHA is caused by a combination of a warm IgG antibody and a cold IgM antibody. The DAT is usually positive for IgG and C3d. Cold-associated symptoms rarely appear, and the cold antibody may have a low antibody titer (eg, <1:64) but with a thermal amplitude up to 30 to 37°C. It can be diagnosed with a DAT positive for IgG and C3d, a cold antibody with a thermal amplitude ≥ 30°C, and an appropriate clinical picture.
Paroxysmal cold hemoglobinuria (PCH) usually occurs in children. The hemolysis can be severe and intravascular but is typically transient following infection. PCH is caused by a biphasic IgG antibody that binds to red cells at low temperature and causes complement-mediated lysis as the temperature is increased. It can be diagnosed in patients with AIHA and a positive Donath-Landsteiner test. Testing should be considered in patients with AIHA and a DAT positive for C3d ± IgG (the DAT is sometimes negative), when CAD has been excluded, and there is hemoglobinuria, cold-associated symptoms, atypical serological features, or patients younger than 18 years old.
Autoantibody specificity in PCH is usually anti-P, in contrast to CAD, in which specificity is usually anti-I, but sometimes it is anti-i or anti-PR. Warm AIHA autoantibodies are usually directed against a high incidence antigen on the red cell surface, but ∼3% have specificity (eg, anti-e). Criteria for the diagnosis of AIHA and its subtypes, as well as definitions of treatment response, have varied among studies. An international consensus on terminology and response criteria would be useful to increase comparability between clinical trials.
Treatment of AIHA
Secondary AIHA
British guidelines on secondary AIHA have recently been published.4 In adults, the most common associations are hematological malignancy, infection, and autoimmune disorders. A broad strategy is to treat the underlying condition according to best practice. Successful treatment may also improve the AIHA, but that is not always the case because the strength of the association varies. If the associated condition does not require treatment, AIHA can usually be approached in a similar fashion to primary AIHA, although treatment decisions must be individualized. For example, when AIHA is associated with some disorders, such as SLE, the burdens and benefits of splenectomy are less favorable compared with primary warm AIHA. Evan’s syndrome, in which AIHA is associated with immune thrombocytopenia (ITP), has important differences from primary AIHA, with regard to the exclusion of alternative causes of a bilineage cytopenia, identifying secondary causes such as autoimmune lymphoproliferative syndrome, and the management of its often relapsing/remitting clinical course.
B-cell malignancies are a common association (eg, AIHA occurs in ∼5-10% of patients with chronic lymphocytic leukemia [CLL]). In patients with active CLL that also requires treatment, combinations, such as rituximab, cyclophosphamide, and dexamethasone (RCD); rituximab, cyclophosphamide, vincristine, and prednisolone (R-CVP); and bendamustine and rituximab (BR), appear to be effective. New targeted therapies, such as obinutuzumab, ibrutinib, idelalisib, and venetoclax, could potentially be beneficial for patients with AIHA; however, vigilance is needed because of potential for development of drug-induced immune cytopenias, as previously observed with single agent fludarabine.5 This issue has also been raised with ibrutinib, but emergent AIHA during ibrutinib therapy appears to be an expression of CLL activity rather than drug-induced hemolysis, and treatment can be administered in patients with a history of AIHA.5,6
General strategies for primary AIHA
All patients
Treat any identified underlying cause, such as infection.
Erythropoiesis increases in response to hemolysis, resulting in greater demand for folate. Folic acid is a well-tolerated supplement to prevent a deficiency that could exacerbate the anemia.
Compatibility testing for patients requiring transfusion focuses on identifying the patient’s ABO CcDEe and K status, as well as the presence of alloantibodies. This can be achieved by special techniques, such as auto- or alloadsorbtion (warm autoantibodies) or prewarming screening cells and patient’s plasma separately to 37°C before use in the indirect anti-globulin test (cold autoantibodies).7 Genotyping can be useful, particularly for complex cases in which it is desirable to match for an extended phenotype. Clinicians should be aware that full compatibility testing can take 4 to 6 hours and that the units provided will not be labeled as “compatible,” because autoantibodies from unmodified patient plasma are likely to bind donor red cells in an indirect anti-globulin test cross-match. Without a history of transfusion or pregnancy, the patient is unlikely to have an alloantibody. If anemia is life threatening, transfusion of ABO Rh and K matched red cells may be preferable to waiting for full-compatibility testing.
Venous thromboembolism prophylaxis: thrombosis risk with hemolysis is still underestimated but remains an important cause of morbidity and mortality in AIHA. In some series, venous thromboembolism events occurred in up to 20% to 25% of patients, usually when hemolysis was active. Larger studies have shown higher rates of venous and arterial thrombosis in patients with AIHA8,9 and, more recently, in those with CAD specifically.10 Perhaps not surprisingly, the risk increases with higher LDH levels (the role of intravascular hemolysis and thrombosis risk has been discussed previously11 ), as well as in patients with previous splenectomy. Thromboprophylaxis with low-molecular-weight heparin is recommended for in-patients with an acute exacerbation of hemolysis and should be considered in ambulatory patients during severe exacerbations (hemoglobin < 85 g/L).12
CAD patients
In CAD patients, intravascular hemolysis can be triggered by bacterial or other febrile illnesses and should be treated promptly.
Patients with CAD should avoid exposure to cold weather where possible and dress to protect the distal extremities.
While in hospital, the patient should be kept warm, especially in the perioperative period. Cardiopulmonary bypass presents a particular challenge related to the usual practice of systemic cooling and cold cardioplegia. However, systemic normothermia and warm cardioplegia is a recognized method, and adjunctive eculizumab has been successfully used to cover bypass surgery.13
Patients should not receive cold fluids IV, and use of an in-line blood warmer is recommended.
Patients receiving steroids
Because of an increased risk for upper gastrointestinal complications, antacid therapy, such as a proton pump inhibitor, is recommended in patients taking steroids, if there is an additional risk factor, such as previous peptic ulcer disease, age ≥ 60 years, concomitant nonsteroidal anti-inflammatory drug/anticoagulant/aspirin use, or concomitant thrombocytopenia.
Fracture risk is increased in patients receiving glucocorticoids for ≥3 months (prednisolone ≥ 7.5 mg/d). It is recommended that these patients are advised about lifestyle modification (smoking cessation, limiting alcohol intake to ≤2 U/d, and engaging in regular weight-bearing exercise) and maintain adequate daily vitamin D (600-800 IU) and calcium (1000-1200 mg) intake through diet, if possible, or supplements, if needed.14 In addition, for adults ≥40 years of age, the initial absolute fracture risk should be estimated using the online tool FRAX (https://www.shef.ac.uk/FRAX/tool.jsp), as well as bone mineral density testing, if available, to determine whether an oral bisphosphonate should also be prescribed.14 However, some guidelines also recommend that men older than 50 years of age and postmenopausal women are at sufficient risk that treatment may be considered without further assessment.15
Primary warm AIHA
Although most patients require treatment, watchful waiting may be appropriate in a minority of patients with mild asymptomatic anemia. For many years, the standard first-line therapy has been glucocorticoids, typically prednis(ol)one at a starting dose of 1 mg/kg. The response rate is ∼80%, but ∼60% of these lose their response upon weaning or discontinuing steroids.16,17 Although AIHA is rare, with a weak evidence base to guide treatment, 2 phase 3 prospective randomized trials have compared predniso(lo)ne with predniso(lo)ne-rituximab as first-line treatment for warm AIHA, although not all cases were primary. A Danish study randomized 64 patients to prednisolone or prednisolone with rituximab (375 mg/m2 weekly for 4 weeks). The response rate to prednisolone-rituximab was significantly higher (75% vs 36%; P = .003) at 12 months, with no difference in adverse reactions.18 A French study randomized 32 patients to prednisone-placebo vs prednisone-rituximab (1 g on days 1 and 15). Again, the primary end point of overall response at 12 months was significantly higher (75% vs 31%; P = .032) in patients receiving rituximab, without excess adverse events.19 Ideally, larger studies of longer duration, powered to determine the impact of rituximab on transfusion requirement, cumulative steroid exposure, and health-related quality of life, would help to inform the first-line standard of care. However, given that steroids are often used to rescue relapsing patients, and prolonged steroid use carries a significant risk for infection, diabetes, and fracture,17,20 the use of first-line rituximab with a relatively short course of steroids has been suggested for elderly patients or those with comorbidities.19
In patients receiving first-line steroids, the long-standing second-line treatment of choice was splenectomy.21 However, recent United Kingdom12 and French22 guidelines have instead favored rituximab. This reflects the relatively large number of published case series that have increased certainty in its effectiveness and tolerability. In a meta-analysis of 154 patients, the overall response rate of warm AIHA to rituximab was 79%, although not all cases were primary, and some received concomitant steroids.23 Adverse events occurred in 38 of 364 (14%) AIHA patients receiving rituximab, including 18 severe infections. Approximately 25% to 50% of responding patients will relapse within 2 to 3, whereas long-term response rates are unknown.12
Approximately 70% of patients with primary warm AIHA respond to splenectomy,12 and this continues to be a useful second-line option when economic restraints limit access to rituximab.24 The infective and thrombotic complications following splenectomy are well recognized. Small series suggest ∼30% relapse in the short term,17,25 but unlike splenectomy for ITP, the long-term durability of remission is unknown. Alternative, relatively nontoxic immunosuppressive therapies, such as mycophenolate mofetil, azathioprine, and cyclosporin, can also be used as third-line treatment. Response rates are difficult to estimate because evidence of efficacy is limited to small series that often lack detail, but they are likely to be lower than with rituximab or splenectomy.
Patients who are steroid sensitive, but refractory or relapsing after third-line therapies, may tolerate longer-term treatment with low-dose predniso(lo)ne (≤10 mg), which can be used in conjunction with a steroid-sparing agent.26 Alternatives with greater potential for toxicity include cyclophosphamide,27,28 alemtuzumab,29 and hematopoietic stem cell transplantation.30
Rescue therapies
Patients can take time to respond to immunosuppressive therapy (eg, median of 3-6 weeks for rituximab for warm AIHA).12 Therefore, those with severe transfusion-dependent hemolysis may need rescue therapy. This may be with steroids if the patient is known to be sensitive. Alternatively, approximately one third of patients will respond to IV Ig, with better responses observed in the most severe cases (hemoglobin ≤ 70 g/L). The clinical severity of AIHA is influenced by the marrow’s compensatory response, and reticulocytopenia has been observed, particularly in children and severe cases, possibly as a result of autoantibodies acting against erythroid precursors.31 This observation provides rationale for the empirical use of recombinant erythropoietin when hemolysis is severe with a poor reticulocyte response; however, parvovirus, hematinic deficiency, and marrow infiltration should also be considered. Emergency splenectomy or partial splenic embolization in unfit patients has been used acutely to treat severe hemolysis.
Autoreactive IgG and IgM can be produced in warm AIHA; when bound to the erythrocyte, both are able to bind C1q and activate the classical complement pathway. C1 esterase inhibitor (C1-INH) is a commercially available inhibitor of the classical pathway licensed for hereditary angioedema with a favorable safety profile. C1-INH has been used to improve response to transfusion in a patient with severe IgM warm AIHA.32 Furthermore, 4 patients with CAD or mixed AIHA and a C3d-positive DAT all responded to the C1-INH BERINERT (20 μg/kg daily for 6-20 days given with prednisolone, with or without rituximab).33 Although data are sparse, C1-INH may have a role in stabilizing severe hemolysis if the DAT is positive for C3d.
Future prospects
Future prospects for primary warm AIHA include inhibition of the following:
B cells with alternative anti-CD20 monoclonal antibodies (mAbs) (eg, obinutuzumab)
Mammalian target of rapamycin (eg, sirolimus)
Proteasomes (eg, bortezomib)
Spleen tyrosine kinase (Syk) (eg, fostamatinib)
Neonatal Fc receptor (FcRn)
Bruton tyrosine kinase (BTK)
Complement pathways
Following the success of rituximab, additional anti-CD20 mAbs were developed, primarily to target B-cell malignancies. These vary in terms of their antibody effector functions, such as complement-dependent cytotoxicity, directly induced nonapoptotic cell death, and FcγR-dependent mechanisms. Third-generation mAbs have been engineered to enhance effector functions (eg, removal of the Fc glycan fucose to increase FcγRIIIA binding affinity; obinutuzumab).34 Other anti-CD20 mAbs are being explored in autoimmune diseases, such as ITP (veltuzumab) and multiple sclerosis (ublituximab), but their role in AIHA awaits investigation. Another novel strategy aiming to reduce relapse would be maintenance treatment with an anti-CD20 mAb.
The mammalian target of rapamycin inhibitor sirolimus, which increases T-regulatory cells and induces apoptosis in abnormal lymphocytes, has been used successfully in children to treat autoimmune lymphoproliferative syndrome, posttransplant AIHA, and 4 of 4 patients with refractory primary AIHA.35
The proteasome inhibitor bortezomib has been used to treat antibody-mediated hematological disorders, including thrombotic thrombocytopenic purpura, acquired hemophilia, and warm AIHA. It causes apoptosis of antibody-producing plasma cells, downregulates NF-κB–mediated proinflammatory signaling, depletes autoreactive T-helper cells, and interferes with antigen presentation.36 Long-lived autoreactive plasma cells have been shown to persist in the spleen of warm AIHA patients who failed to respond to rituximab.37 In a retrospective study, 3 of 4 refractory patients with warm AIHA achieved at least a partial response to bortezomib, although all patients were receiving concomitant therapies, and prospective studies are needed.36
Binding of FcγR on inflammatory cells causes activation of Syk, which leads to phagocytosis and immune activation. The Syk inhibitor fostamatinib achieved a stable response in 18% of ITP patients treated in 2 phase 3 randomized controlled trials,38 and in the interim report of a phase 2 trial in warm AIHA (ClinicalTrials.gov identifier NCT02612558), 9 of 17 (53%) patients responded to fostamatinib (150 mg, twice daily).39
FcRn is a widely expressed receptor that is able to bind and transport IgG antibodies within and between cells. Endosomal FcRn prolongs the half-life of IgG by recycling antibody that would otherwise undergo lysosomal degradation. FcRn inhibitors are being studied in a variety of autoimmune conditions with the aim of increasing catabolism of pathogenic IgG. A phase 1 study of the FcRn inhibitor SYNT001 in warm AIHA is underway (NCT03075878).
BTK is a signaling element downstream of the B-cell and Fcγ receptors. The oral BTK inhibitor PRN1008 is being studied in ITP (NCT03395210), but it might also be applicable to AIHA.
APL-2, an inhibitor of C3, is being evaluated in an open-label phase 2 study of patients with warm AIHA and CAD (NCT03226678).
Primary CAD
Patients with mild asymptomatic anemia can be monitored without specific treatment, which should be reserved for symptomatic anemia, severe circulatory symptoms, or transfusion dependence. Because exacerbations can be transient, a period of support with transfusion may be useful before determining the need for pharmacological intervention.
First-line treatment is usually rituximab (375 mg/m2 weekly for 4 weeks). Approximately 50% responded in 2 prospective case series, but complete responses were rare.3 This lower response rate compared with warm AIHA may be due to the presence of an underlying clonal lymphoproliferative disorder and/or because the response criteria used required improvement in cold associated symptoms and the clonal disease, as well as hemolysis. Unfortunately, the median response duration was only 11 months (Table 3), although 6 of 10 responding patients also responded to a second course.40
Comparison of Nordic studies of rituximab and rituximab combination therapies for CAD
| BR (n = 45)45 | Fludarabine-rituximab (n = 29)46 | Rituximab (n = 27)40 | |
|---|---|---|---|
| Regimen | Rituximab: 375 mg/m2 day 1 Bendamustine: 90 mg/m2 days 1 and 2, 4 cycles at a 28-d interval | Rituximab: 375 mg/m2 day 1 Fludarabine (oral): 40 mg/m2 days 1 to 5, 4 cycles at a 28-d interval | Rituximab: 375 mg/m2 day 1 (weekly for 4 wk) |
| Response rate, % | |||
| Overall response | 71 | 76 | 52 |
| Complete response | 40 | 21 | 4 |
| Median time to response (range), mo | 1.9 (0.25-12) | 4.0 (0.3-6.0) | 1.5 (0.5-4.0)* |
| Sustained response | |||
| % | 91% at median FU 36 mo | 77% at median FU 33 mo | Not reported |
| Median response duration | Observed = 32 mo | Estimated > 66 mo | Observed = 11 mo (range 2-42)* |
| Tolerance | 33% G3 or G4 neutropenia | 41% G3 or G4 hematologic toxicity | 3% G4 hematologic toxicity |
| 11% infections | 59% G1 to G3 infections | 3% G1 infection* | |
| 1 fatal pneumonia at 5 mo | 1 fatal pneumonia at 9 mo | ||
| 1 fatal episode of hypothermia during rituximab |
| BR (n = 45)45 | Fludarabine-rituximab (n = 29)46 | Rituximab (n = 27)40 | |
|---|---|---|---|
| Regimen | Rituximab: 375 mg/m2 day 1 Bendamustine: 90 mg/m2 days 1 and 2, 4 cycles at a 28-d interval | Rituximab: 375 mg/m2 day 1 Fludarabine (oral): 40 mg/m2 days 1 to 5, 4 cycles at a 28-d interval | Rituximab: 375 mg/m2 day 1 (weekly for 4 wk) |
| Response rate, % | |||
| Overall response | 71 | 76 | 52 |
| Complete response | 40 | 21 | 4 |
| Median time to response (range), mo | 1.9 (0.25-12) | 4.0 (0.3-6.0) | 1.5 (0.5-4.0)* |
| Sustained response | |||
| % | 91% at median FU 36 mo | 77% at median FU 33 mo | Not reported |
| Median response duration | Observed = 32 mo | Estimated > 66 mo | Observed = 11 mo (range 2-42)* |
| Tolerance | 33% G3 or G4 neutropenia | 41% G3 or G4 hematologic toxicity | 3% G4 hematologic toxicity |
| 11% infections | 59% G1 to G3 infections | 3% G1 infection* | |
| 1 fatal pneumonia at 5 mo | 1 fatal pneumonia at 9 mo | ||
| 1 fatal episode of hypothermia during rituximab |
Response criteria for all 3 studies were the same.
FU, follow-up; G, grade.
Values are calculated from 37 treatment episodes in 27 patients.
Rituximab has been combined with bendamustine (BR) or fludarabine in 2 prospective uncontrolled studies (Table 3). Although the studies are not directly comparable, the response criteria used were the same, and there appeared to be a higher rate of complete remission and fewer infections in patients receiving BR. Combination therapy provides more opportunity for a durable response but with a greater risk for toxicity. BR is a suitable second-line option for fit patients or a first-line option in selected patients with severe disease.
A prospective open-label phase 2 study (NCT01696474) reported on 19 previously treated CAD patients who received a single course of bortezomib (1.3 mg/m2) on days 1, 4, 8, and 11.41 Improved hemolysis was documented in 6 of 19 patients, including complete resolution in 3. Response was sustained in 4 of 6 patients at a median of 16 months (range, 10-31). Some activity against the underlying B-cell clone was observed, with a <50% decrease in monoclonal Ig from pretreatment levels in 3 of 13 evaluable patients and a reduction in marrow B-cell lymphoid infiltrate >50% in 2 of 5 cases. Although further studies are needed, therapeutic options for CAD are limited, and bortezomib may be a suitable second-line option in patients who are not fit for combination therapy, such as BR or rituximab plus fludarabine.3
Emergency treatment
CAD patients usually take several months to respond to rituximab-based therapy (Table 3), whereas hemolysis can be severe and intravascular. In addition to blood transfusion, daily or alternative-day plasma exchange of 1 to 1.5 times the plasma volume with albumin has been used as a bridging therapy, although evidence is mixed and largely confined to case reports. Although not recommended for routine treatment of CAD, steroids may have a limited role as rescue therapy in severe cases. Patients with acute severe intravascular hemolysis may also respond to the C5 inhibitor eculizumab, which blocks the terminal complement pathway. Its activity in CAD is supported by case reports and a prospective study of 13 patients who received 6 months of treatment, with a reduction in transfusion requirements and LDH.42
Future prospects
Given the mechanism of hemolysis outlined previously, inhibition of the classical complement pathway is a rational and promising therapeutic approach for CAD. Treatment would need to be maintained to have a continuing effect and is not expected to improve cold-associated symptoms, which result from agglutination of antibody-bound red cells upstream of complement activation. In a phase 1b study, 6 CAD patients received a 10 mg/kg loading dose of the anti-C1s antibody BIVV009 (sutimlimab) and then received 60 mg/kg 1 to 4 days later, followed by 3 weekly doses of 60 mg/kg. Treatment was well tolerated, and all 6 patients responded with a mean increase in hemoglobin of 43 g/L. On-going responses were observed in a named patient program using a fixed biweekly dose of 5.5 g with up to 18 months of observation.43 Phase 3 CAD clinical trials (NCT03347396, NCT03347422) are now open.
The C3 inhibitor APL2 is being evaluated in warm AIHA and CAD, as previously discussed.
Many more complement inhibitors are under development for various clinical indications and have been recently reviewed.44
Given the clonal nature of CAD, newer targeted therapies for B-cell malignancies, such as the BTK inhibitor ibrutinib and B-cell receptor pathway–modulating drugs (eg, idelalisib and venetoclax) might have activity in CAD, although the hypothesis awaits investigation.3
Correspondence
Anita Hill, Department of Haematology, St James's University Hospital, Beckett St, Leeds LS9 7TF, United Kingdom; anitahill@nhs.net.
