

Abstract
During developmental hematopoiesis, multilineage hematopoietic progenitors are thought to derive from a subset of vascular endothelium. Herein, we define the phenotype of such hemogenic endothelial cells and demonstrate, on a clonal level, that they exhibit multilineage hematopoietic potential. Furthermore, we have begun to define the molecular signals that regulate their development. We found that the formation of yolk sac hemogenic endothelium and its hematopoietic potential were significantly impaired in the absence of retinoic acid (RA) signaling, and could be restored in RA-deficient (Raldh2−/−) embryos by provision of exogenous RA in utero. Thus, we identify a novel, critical role for RA signaling in the development of hemogenic endothelium that contributes to definitive hematopoiesis.
Introduction
In both mouse and human, the first blood and endothelial cells derive from Flk1+ yolk sac mesoderm in response to signals produced by the adjacent visceral endoderm.1-3 Between embryonic day (E) 7.0 and E7.25, primitive hematopoietic cells emerge in the developing blood islands of the yolk sac (Figure 1) comprising a heterogeneous population of myeloid and nucleated erythroid progenitors.4,5 As maturing blood islands coalesce, vascular channels are formed throughout the yolk sac, and the developing heart tube establishes plasma flow within these channels.6 Shortly thereafter, immature erythroblasts enter the plasma circulation. This initial burst of hematopoietic activity is followed by a second wave of hematopoiesis, established first in the yolk sac where multipotential (definitive) blood progenitors become detectable at about E8.25.5 In the embryo proper, definitive hematopoietic progenitors first arise around E10.5 (E30-40 in humans) in the aorta-gonad-mesonephros (AGM) region.7,8

Increasing evidence suggests that during embryonic vasculogenesis, multipotent hematopoietic progenitor cells derive from vascular endothelium in both the yolk sac and embryo proper. In the chick, mouse and human, developing clusters of CD45+ hematopoietic progenitors are found to arise in direct physical association with vascular endothelial cells of the dorsal aorta, and umbilical and vitelline vessels.9-11 In the adult, hematopoietic stem cells and early progenitors have similarly been shown to reside in direct contact with sinusoidal endothelium of the bone marrow niche.12 Stepwise emergence of the hematopoietic lineage from endothelial-like precursors has been demonstrated by in vitro studies in which Flk1+ cells derived from either murine embryonic stem cells or E7.5-E9.5 yolk sac were shown to generate multipotent hematopoietic progenitor cells via a VE-cadherin+ intermediate.13,14 Thus, it is proposed that definitive hematopoiesis requires a coordinated developmental transition in which multilineage hematopoietic progenitors derive from a specialized subset of vascular endothelium. However, such specialized endothelial cells, derived from in vivo sites of definitive hematopoiesis, have not previously been shown to exhibit multilineage hematopoietic potential on a single-cell level.
We have previously identified and characterized a population of yolk sac and embryonic vascular endothelial cells with hemogenic potential.15 These cells are Flk1+ and CD45−, and are phenotypically distinguished from other Flk1+CD45− endothelial cells by their expression of c-Kit, and Hoechst dye-efflux capacity (known as “side population” [SP] phenotype). From the time of definitive hematopoietic induction in the yolk sac at approximately E8.25, the Flk1+c-Kit+CD45− subset of the yolk sac SP differentiates through a VEcad+ SP intermediate into Flk1−c-Kit+CD45+ SP cells, which demonstrate multilineage hematopoietic colony-forming activity in vitro.15 In our current studies, we demonstrate on a clonal level using a novel in vitro assay, that yolk sac Flk1+c-Kit+CD45− SP cells represent specialized endothelial cells with multilineage hematopoietic potential (hemogenic endothelium). In addition, we have begun to define the molecular signaling pathway that induces the hemogenic potential of a subset of primordial endothelium during vasculogenesis.
Of the many factors known to regulate vasculogenesis in vivo, retinoic acid (RA) has been shown to play an especially important role in regulating endothelial cell development.16,17 RA synthesis occurs via retinaldehyde dehydrogenase-2 (RALDH2) conversion of dietary retinol (vitamin A) into its biologically active metabolite, RA, in the visceral endoderm. RA receptors alpha-1 and -2 (RARα1/2) are expressed specifically by developing endothelium,16 and embryos lacking RALDH2 activity exhibit abnormal endothelial cell development.16,17 Studies in the mouse suggest that RA also regulates developmental hematopoiesis18,19 ; however, a specific cellular function for RA therein has not yet been described. Here we demonstrate that RA signaling is required for the formation of hemogenic endothelial cells (Flk1+c-Kit+CD45− SP cells) from primordial (nonspecialized) endothelium, and for their multilineage hematopoietic potential in vivo. Thus, we have identified, isolated and characterized hemogenic endothelial cells on a clonal level and have begun to define the molecular pathway regulating their formation and function.
Methods
Tissue processing and genotyping
Endogenous expression of the murine Raldh2 gene was disrupted in CD1 mice as previously described,20 and maintenance of Raldh2+/− heterozygote breeding pairs achieved by sibling mating. Timed pregnancies were obtained by paired matings; morning of detection of a vaginal plug was considered E0.5. All viable offspring were genotyped by polymerase chain reaction (PCR) using gene-specific primers flanking the murine Raldh2 gene locus20 and genomic DNA isolated from tail clippings using the Qiagen DNeasy Tissue Kit (Qiagen Sciences, Germantown, MD). Genotyping of embryos was performed on whole tissue lysates prepared by digestion of embryonic tissue in 50 mM NaOH at 95°C for 15 minutes, and neutralized with 100 mM Tris-Cl (pH 8.0). Genotyping PCR was performed using Platinum Pfx DNA polymerase (Invitrogen, Grand Island, NY) in the following cycling protocol: 1 cycle of 95°C for 2 minutes; 35 cycles of 95°C for 30 seconds, 58°C for 30 seconds, 72°C for 1 minute; followed by 1 cycle of 72°C for 5 minutes.
Reverse transcription and real-time quantitative PCR
Quantitative analysis of gene expression was performed using TaqMan Gene Expression Assay primer-probe sets from Applied Biosystems (Branchburg, NJ). Freshly isolated yolk sac tissues were individually genotyped, then subjected to fluorescence-activated cell sorting (FACS) for cell isolation or pooled for RNA isolation using the Qiagen RNeasy Mini Tissue Kit (Qiagen Sciences). For qPCR, 1 μg total RNA was reverse-transcribed at 42°C for 50 minutes using the SuperScript II First Strand Synthesis Kit (Invitrogen). Real-time PCR was performed using 1/20th of the reverse transcription reaction product, together with TaqMan Universal PCR Reaction Mix (Applied Biosystems). All gene expression assays were performed in triplicate on at least 2 independently isolated RNA samples, and the data were normalized to endogenous β-actin expression.
Immunohistochemistry
For cryosectioning, embryos were fixed in 4% paraformaldehyde (PFA) for 2 hours at 4°C, cryoprotected by 5% to 20% wt/vol sucrose gradient infusion, then embedded in OCT compound (Sakura Finetek USA, Torrance CA) and sectioned at 10 μm. Cryosections were blocked for 1 hour at room temperature (RT) with 10% normal donkey serum (NDS)/0.5% bovine serum albumin (BSA)/phosphate-buffered saline (PBS). Immunohistochemistry was performed according to standard methods using rabbit anti–mouse RARα (1:100; BioMol, Plymouth Meeting, PA) and goat anti–mouse c-Kit (1:100; R&D Systems, Minneapolis, MN) antibodies diluted in 10% NDS/0.5% BSA/PBS, and incubated for 4 hours at RT. Sections were incubated for 1 hour at RT with donkey anti–goat Alexa-488 and anti–rabbit Alexa-594 secondary antibodies (Molecular Probes, Carlsbad, CA) diluted 1:500 in 10% NDS/0.5% BSA/PBS, then mounted in VectaShield with DAPI (Vector Laboratories, Burlingame, CA) for microscopic imaging and analysis. Images were captured with a Zeiss AxioVert 200M microscope and AxioCam MRM camera, using Zeiss AxioVision version 4.0 software (Carl Zeiss MicroImaging, Thornwood, NY).
For whole mount immunostaining, yolk sacs were fixed as previously described, then blocked and stained for 12 to 16 hours with rat anti–mouse Flk1 (1:200; BD Pharmingen, San Diego CA), goat anti–mouse c-Kit (1:100; R&D Systems), and rabbit anti–mouse RARα (1:100; BioMol) antibodies diluted in 5% NDS/2% BSA/PBS. Yolk sacs were incubated for 2 hours at RT with donkey anti–rat Alexa-488, anti–goat Alexa-594, and anti–rabbit Alexa-633 secondary antibodies (1:500; Molecular Probes), then flat-mounted in VectaShield with DAPI for microscopic imaging and analysis. Images were captured with a Zeiss LSM 510 META confocal inverted microscope using 20× 0.8 NA objective and LSMII imaging software (Carl Zeiss MicroImaging).
For benzidine staining of hemoglobinized cells, unfixed embryos were incubated in 12% glacial acetic acid/4% benzidine/0.3% H2O2 for 10 minutes, cleared in methanol, and photographed immediately for analysis. Images were captured using a Zeiss Stemi 2000-CS dissecting microscope and color camera, with Zeiss AxioCam version 1.1.6 software (Carl Zeiss MicroImaging). All photographic images were taken at 10× magnification and analyzed after acquisition using Adobe Photoshop CS (Adobe Systems, San Jose, CA).
Flow cytometry and FACS
Hoechst staining of yolk sac tissues for isolation of SP cells was performed using a modification of standard published methods.21 Yolk sacs were individually genotyped, pooled, and digested with 0.05% collagenase type II (Sigma-Aldrich, St Louis, MO) in HBSS/10% FBS for 30 minutes at 37°C. Tissue digests were homogenized by trituration with a 26-g needle and syringe and stained with 5 μg/mL Hoechst 33 342 (Sigma-Aldrich) for 1 hour at 37°C. Cells were subsequently stained with various combinations of the following rat anti–mouse secondary antibody conjugates: Flk1-phycoerythrin (PE), CD45-fluorescein isothiocyanate (FITC), CD31-FITC, CD41-FITC, Ter119-E, and c-Kit-allophycocyanin (APC, 1:100; BD Pharmingen). For detection of VE-cadherin on hemogenic endothelium, cells were first incubated with rat anti–mouse VE-cadherin monoclonal antibodies (1:100; BD Pharmingen), followed by donkey anti–rat Alexa-633 IgG secondary antibodies (Vector Laboratories). Cells were then washed to remove residual anti–rat IgG and secondarily incubated with c-Kit and Flk1 direct antibody conjugates. After immunostaining, all cell preparations were washed and resuspended in Hanks balanced salt solution (HBSS)/10% fetal bovine serum (FBS) with 10 μg/mL propidium iodide (PI; Sigma-Aldrich). All flow cytometry FACS were performed using either a MoFlo (Dako Cytomation, Fort Collins, CO) or FACSAria (BD Biosciences, San Jose, CA) cell sorter.
Methylcellulose hematopoietic colony-forming assay
To assess the hematopoietic potential of phenotypically distinct yolk sac cell populations, individually genotyped tissues were pooled, collagenase-digested, and stained with a combination of Hoechst 33 342 and mouse-specific antibodies as previously described. Individual cell populations were isolated by FACS and sorted directly into 500 μL of MethoCult GF M3434 hematopoietic culture medium (StemCell Technologies, Vancouver, BC) in 24-well culture plates. For single-cell clonal analysis of hemogenic endothelium (Flk1+c-Kit+CD45− SP cells), single cells were sorted into 100 μL M3434 in 96-well fibronectin-coated culture plates. All cultures were incubated at 37°C with 5% CO2, and scored for colony formation at days 1, 3, 5, 7, and 14 as appropriate.
DiI AcLDL endothelial cell assay
To demonstrate endothelial phenotypic function in cultures of yolk sac–derived hemogenic endothelium, cells were assayed for active uptake of DiI AcLDL. Individual Flk1+cKit+CD45− SP cells isolated from wild-type (WT) E9.5 mouse yolk sac were plated in fibronectin-coated 96-well culture dishes (BD Biosciences, Bedford MA) with 100 μL of MethoCult GF M3434 medium (StemCell Technologies). On day 1 of culture, cells were stained with 10 μg/mL 1,1′-dioctadecyl-3,3,3′-tetramethyl-indocarbocyanine–labeled acetylated low density lipoprotein (DiI AcLDL; Molecular Probes) for 4 hours at 37°C, then visualized by fluorescence microscopy using a 594-nm laser filter for detection of DiI AcLDL.
In vivo phenotypic rescue studies
Phenotypic rescue of Raldh2−/− embryos in utero was achieved by supplementing the food supply of pregnant dams with all-trans retinoic acid (ATRA; Sigma-Aldrich). Beginning at E7.5, all food provided to pregnant Raldh2+/− dams was supplemented with 100 μg/mL ATRA, combined at a ratio of 100 μg ATRA/g normal rodent chow, and continued for 48 hours until the time of sacrifice at E9.5, as described.17 All embryos obtained from ATRA-supplemented pregnancies were genotyped as previously described, before subsequent analysis.
Fluorescence recovery after photobleaching
Embryos from E8.0 pregnant Raldh2+/− dams were dissected at 37°C in 90% DMEM/F12 supplemented with 8% FBS and 2% penicillin/streptomycin. Using a pulled quartz needle, nanoliter volumes of 50 mg/mL 10 000 MW fluorescein dextran (Molecular Probes) were then injected into the heart tube of 6- to 8-somite stage embryos. Embryos were transferred into dissection media with 50% normal rat serum, and allowed to recover at 37°C with 5% CO2 for 30 minutes. Individual embryos were then transferred to the wells of a Lab-Tek culture chamber (Nalgene Nunc, Rochester, NY), and placed on the preheated stage of a Zeiss LSM 510 META confocal inverted microscope for real-time imaging of the caudal yolk sac arterial vessels. After verification of dextran distribution through the vascular plexus, a region of interest was defined within a plexus vessel. Scan speed was set at 45 ms per frame, laser power 2%. Using the LSM software bleach function, each yolk sac was continuously imaged prior to and spanning approximately 800 frames, after a 50-frame 100% laser power bleach event. Measurements were obtained at 3 independent locations within the yolk sac near the caudal aspect of each embryo, which were subsequently genotyped as previously described.
Mean fluorescence with respect to time was tabulated and the recovery curve fitted to the
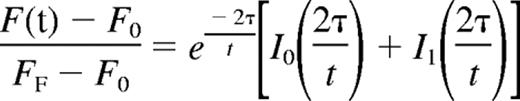
where F(t) indicates the fluorescence intensity, F0 the initial postbleach fluorescence intensity, FF the final level of fluorescence recovery, I0 and I1 the zero- and first-order Bessel functions, and τ the characteristic diffusion time.22 The diffusion coefficient was defined as D = d2/4τ, where d is the diameter of the region of interest (ROI).22 The baseline for pure diffusion was obtained by injecting embryos with fluorescein-dextran as described.6 Statistical significance of the average diffusion coefficients for WT and Raldh2−/− mutant embryos was determined by independent Student t test using a confidence interval of 95% (P ≤ .05). All research involving mice was reviewed and approved by the Baylor College of Medicine Animal Care and Use Committee. This study was conducted in accordance with the Declaration of Helsinki.
Results
RA signaling is not required for primitive hematopoiesis
Primitive hematopoiesis is marked by the emergence of blood islands in the yolk sac between E7.0 and E7.5 and subsequent entry of nucleated erythroblasts into the circulation. Quantitative measurement of Ter119 and CD41 expression in 6- to 8-somite WT and Raldh2−/− yolk sacs by FACS revealed no impairment of primitive erythroid development before the onset of systemic blood circulation (Figure 2A). Benzidine staining for hemoglobinized erythroblasts of the yolk sac blood islands also confirmed normal initiation of primitive hematopoiesis in RA-deficient embryos at this time point (Figure 2B).

RA signaling is not required for primitive hematopoiesis. (A,C) FACS analysis of the primitive hematoendothelial compartment in E8.0 Raldh2−/− and WT yolk sac. All population data were calculated as a percentage of the total live cell population plus or minus SEM (n ≥ 3). Statistical analysis of significance was determined by Student t test, with a confidence interval of 95% (P ≤ .05). (B) Benzidine staining reveals normal blood island development in E8.0 Raldh2−/− and WT embryos. (D) qPCR analysis of hematopoietic gene expression in E8.0 Raldh2−/− versus WT littermate yolk sacs. All data points were calculated as gene expression relative to endogenous β-actin expression (n = 3). (E) Quantitative measurement of intravascular plasma flow in precirculation E8.0 Raldh2−/− and WT embryos. FRAP was used to determine the average rate of plasma flow in caudal vessels of 6- to 8-somite Raldh2−/− (n = 12) and WT (n = 63) yolk sacs, before entry of blood cells into the systemic circulation. Plasma flow was calculated as the mean per embryo plus or minus SEM.
RA signaling is not required for primitive hematopoiesis. (A,C) FACS analysis of the primitive hematoendothelial compartment in E8.0 Raldh2−/− and WT yolk sac. All population data were calculated as a percentage of the total live cell population plus or minus SEM (n ≥ 3). Statistical analysis of significance was determined by Student t test, with a confidence interval of 95% (P ≤ .05). (B) Benzidine staining reveals normal blood island development in E8.0 Raldh2−/− and WT embryos. (D) qPCR analysis of hematopoietic gene expression in E8.0 Raldh2−/− versus WT littermate yolk sacs. All data points were calculated as gene expression relative to endogenous β-actin expression (n = 3). (E) Quantitative measurement of intravascular plasma flow in precirculation E8.0 Raldh2−/− and WT embryos. FRAP was used to determine the average rate of plasma flow in caudal vessels of 6- to 8-somite Raldh2−/− (n = 12) and WT (n = 63) yolk sacs, before entry of blood cells into the systemic circulation. Plasma flow was calculated as the mean per embryo plus or minus SEM.
We previously determined that SP cells of the E8.0 yolk sac represent primordial endothelium (Flk1+CD31+CD45−), which gives rise to hemogenic endothelium, and later hematopoietic progenitors.15 Thus, we aimed to determine whether the formation of this hemogenic endothelial precursor population was disrupted in the absence of RA signaling. We quantified the number of yolk sac SP cells in WT and Raldh2−/− mutants before the onset of hemogenic endothelial cell formation, and found no quantitative differences (Figure 2C). We also saw no quantitative difference in the expression of hematopoietic genes in this population of cells in Raldh2−/− yolk sacs compared with WT littermates (Figure 2D). These findings are consistent with normal formation of the hemogenic endothelial cell precursor population of the yolk sac in the absence of RA signaling.
We next aimed to determine whether hemodynamic flow, which is known to regulate endothelial cell phenotype6,23 and thus may modulate the specialization of primordial endothelium to hemogenic endothelium, was disrupted in Raldh2−/− mutants. Measuring the rate of plasma flow within yolk sac vascular channels via heart tube injection of FITC-conjugated dextran and fluorescence recovery after photobleaching (FRAP) analysis, we found no difference between E8.0 WT and Raldh2−/− mutants (Figure 2E). Collectively, our data indicate that early endothelial cell development, primitive erythroblast formation, and the onset of plasma circulation were all unaffected by loss of RA signaling in vivo. Thus primitive hematopoiesis is not controlled by RA signaling, and the defects in blood production in Raldh2−/− mutants are likely due to impairment of definitive hematopoiesis, to which hemogenic endothelium contributes.
Yolk sac Flk1+c-Kit+CD45− SP cells are endothelial cells with hematopoietic potential
Hemogenic endothelium with multilineage hematopoietic potential emerges at approximately E8.25 in the yolk sac as Flk1+c-Kit+CD45− SP cells.15 Using whole mount immunostaining of E9.5 yolk sac to colocalize Flk1 and c-Kit expression, we found Flk1+c-Kit+ cells distributed throughout the yolk sac capillaries, particularly on the arterial side of the yolk sac vasculature (Figure 3A). The greatest concentration of hemogenic endothelial cells, however, was found within the lumenal wall of the yolk sac vitelline vein (Figure 3Aiv), consistent with previous reports suggesting the emergence of hemogenic endothelium within the embryonic umbilical and vitelline vessels.24,25

Yolk sac Flk1+c-Kit+CD45− SP cells are endothelial cells with hematopoietic potential. (A) Whole mount immunohistochemical colocalization of (i) Flk1 (Alexa-488; green) and (ii) c-Kit (Alexa-594; red) on hemogenic endothelial cells of the E9.5 murine yolk sac arterial capillary bed. (Aiii,iv) Color-merged confocal z-stack projection of E9.5 yolk sac whole mount immunostain. White arrowheads indicate Flk1+c-Kit+ hemogenic endothelial cells localized to the vitelline vein. (B) Clonal analysis of Flk1+c-Kit+CD45− SP hemogenic endothelial cells cultured on fibronectin with MethoCult GF M3434. (Bi) Day 1 culture, single Flk1+c-Kit+CD45− SP cell stained for 4 hours at 37°C with 10 μg/mL DiI AcLDL. (Bii) Day 3 culture showing adherent cobblestone morphology of proliferating Flk1+c-Kit+CD45− SP hemogenic endothelial cell colony. (Biii) Day 7 culture showing emergence of multipotent HPC from clonal Flk1+c-Kit+CD45− SP hemogenic endothelial cell culture. White arrowheads indicate representative cells displaying characteristic endothelial cell morphology; black arrowheads indicate hematopoietic progenitor morphology. (C) Multilineage hematopoietic potential of Flk1+c-Kit+CD45− SP hemogenic endothelial cell clones cultured on fibronectin in MethoCult GF M3434, (Ci) BFU-E erythroid colony, (Cii) CFU-GM granulocyte-monocyte colony, (Ciii) CFU-GEMM multipotent hematopoietic progenitor colony. (D) Immunohistochemical colocalization of c-Kit (Alexa-488; green) and RARα (Alexa-594; red) on hemogenic endothelial cells of the E9.5 murine yolk sac. White arrowheads indicate c-Kit+RARα+ endothelial and hematopoietic cells. Scale bars in all panels represent 100 μm (20× magnification).
Yolk sac Flk1+c-Kit+CD45− SP cells are endothelial cells with hematopoietic potential. (A) Whole mount immunohistochemical colocalization of (i) Flk1 (Alexa-488; green) and (ii) c-Kit (Alexa-594; red) on hemogenic endothelial cells of the E9.5 murine yolk sac arterial capillary bed. (Aiii,iv) Color-merged confocal z-stack projection of E9.5 yolk sac whole mount immunostain. White arrowheads indicate Flk1+c-Kit+ hemogenic endothelial cells localized to the vitelline vein. (B) Clonal analysis of Flk1+c-Kit+CD45− SP hemogenic endothelial cells cultured on fibronectin with MethoCult GF M3434. (Bi) Day 1 culture, single Flk1+c-Kit+CD45− SP cell stained for 4 hours at 37°C with 10 μg/mL DiI AcLDL. (Bii) Day 3 culture showing adherent cobblestone morphology of proliferating Flk1+c-Kit+CD45− SP hemogenic endothelial cell colony. (Biii) Day 7 culture showing emergence of multipotent HPC from clonal Flk1+c-Kit+CD45− SP hemogenic endothelial cell culture. White arrowheads indicate representative cells displaying characteristic endothelial cell morphology; black arrowheads indicate hematopoietic progenitor morphology. (C) Multilineage hematopoietic potential of Flk1+c-Kit+CD45− SP hemogenic endothelial cell clones cultured on fibronectin in MethoCult GF M3434, (Ci) BFU-E erythroid colony, (Cii) CFU-GM granulocyte-monocyte colony, (Ciii) CFU-GEMM multipotent hematopoietic progenitor colony. (D) Immunohistochemical colocalization of c-Kit (Alexa-488; green) and RARα (Alexa-594; red) on hemogenic endothelial cells of the E9.5 murine yolk sac. White arrowheads indicate c-Kit+RARα+ endothelial and hematopoietic cells. Scale bars in all panels represent 100 μm (20× magnification).
A subset of hemogenic endothelial cells (∼30%, data not shown) also express the endothelial marker VE-cadherin, which has been shown to mark the transitional phenotype from Flk1+CD45− endothelial cells to Flk1−CD45+ hematopoietic progenitors.14 To determine whether definitive hematopoietic potential is enriched within and/or restricted to the VE-cadherin+ hemogenic endothelial population, we used FACS to isolate Flk1+c-Kit+CD45− SP cells from E9.5 yolk sac, then further separated them into VE-cadherin–positive and –negative subpopulations. Cells from each fraction were cultured in both MethoCult GF M3434 and on fibronectin-coated plates in EGM2 endothelial medium, for up to 14 days. Multipotent hematopoietic colony-forming activity was observed in both Flk1+c-Kit+VEcad+ SP and Flk1+c-Kit+VEcad− SP hematopoietic cultures (data not shown), and both populations adopted adherent cobblestone/monolayer morphology in EGM2 culture. These data indicate that although a likely marker of the transitional phenotype from Flk1+CD45− hemogenic endothelial cells to Flk1−CD45+ hematopoietic progenitors, expression of VE-cadherin is neither a defining characteristic of the earliest yolk sac–derived hemogenic endothelium nor a prerequisite for its function.
To further demonstrate that Flk1+c-Kit+CD45− SP cells are endothelial cells with multilineage hematopoetic potential, we performed clonal assays in which single Flk1+c-Kit+CD45−VEcad− SP cells were individually cultured in MethoCult GF M3434 on fibronectin-coated plates. After 1 day of culture, all plated cells were adherent, displayed characteristic endothelial cell morphology, and exhibited uptake of DiI AcLDL (Figure 3Bi). By day 3, endothelial colonies had formed (Figure 3Bii), and by days 7 through 14, newly formed clusters of hematopoietic progenitors were observed growing in direct contact with the underlying endothelial cell monolayer in 100% of proliferating cultures (Figure 3Biii). Using this culture system, proliferating cultures were routinely obtained from approximately 20% of all singly plated hemogenic endothelial cells. Morphologic and FACS analysis of colonies formed from the Flk1+c-Kit+CD45− SP cells revealed both multilineage (CFU-GEMM) and lineage-restricted hematopoietic colonies (BFU-E, CFU-GM; Figure 3C); thus confirming, on a clonal level, their specialized ability to generate multipotent hematopoietic progenitors.
Hemogenic endothelial cells express RA receptors
To determine whether the lack of blood production in Raldh2−/− mutants could be due to lack of RA signaling within hemogenic endothelial cells, we examined the expression of RARα1/2 in Flk1+c-Kit+CD45− cells. Immunohistochemical staining of E9.5 yolk sac sections confirmed localization of RARα1/2 in c-Kit+ endothelium, consistent with our previous findings that endothelial cells specifically express RARα1/216 (Figure 3D), whereas FACS analysis confirmed expression of RARα1/2 on approximately 80% of Flk1+c-Kit+CD45− SP cells (data not shown). Thus, the majority of hemogenic endothelial cells have the ability to respond to RA signaling, which may directly regulate their development and hematopoietic function.
RA signaling regulates the differentiation of hemogenic endothelium
To determine whether hemogenic endothelial cells are properly formed from vascular endothelium in the absence of RA signaling, we used Hoechst staining and FACS to isolate and compare 4 previously defined cell populations from E9.5 WT and RA-deficient Raldh2−/− yolk sacs: hemogenic endothelium (Flk1+c-Kit+CD45− SP), multipotent hematopoietic progenitors (Flk1−c-Kit+CD45+ SP), endothelial cells (Flk1+c-Kit−CD45− non-SP), and committed blood progenitors (Flk1−c-Kit+CD45+ non-SP).15 We detected a statistically significant decrease in the total number of SP cells in RA-deficient mutants relative to WT (0.2% vs 0.6%, P = .005; Figure 4A,B). Furthermore, we observed a 10-fold decrease in hemogenic endothelial cells in Raldh2−/− mutants compared with WT littermates (0.01% vs 0.1%, P = .02; Figure 4C). As expected, decreased generation of hemogenic endothelium was associated with loss of multipotent hematopoietic progenitor cells in Raldh2−/− mutants (0.01% vs 0.1% in WT, P = .03; Figure 4C).

RA signaling regulates the development of hemogenic endothelium during murine embryogenesis. (A) Phenotypic characterization of the SP compartment in E9.5 WT, and (B) Raldh2−/− yolk sac. Yolk sacs were stained with Hoechst 33 342, c-Kit-APC, Flk1-PE and CD45-FITC antibodies for visualization and phenotypic analysis of the SP compartment by flow cytometry. Cells were additionally stained with PI to enable exclusion of nonviable cells. (C) Quantitative analysis of the SP compartment in E9.5 Raldh2−/− versus WT yolk sac, calculated as a percentage of the total live cell population plus or minus SEM (n ≥ 3). Statistical analysis of significance was determined by Student t test, with a confidence interval of 95% (P ≤ .05).
RA signaling regulates the development of hemogenic endothelium during murine embryogenesis. (A) Phenotypic characterization of the SP compartment in E9.5 WT, and (B) Raldh2−/− yolk sac. Yolk sacs were stained with Hoechst 33 342, c-Kit-APC, Flk1-PE and CD45-FITC antibodies for visualization and phenotypic analysis of the SP compartment by flow cytometry. Cells were additionally stained with PI to enable exclusion of nonviable cells. (C) Quantitative analysis of the SP compartment in E9.5 Raldh2−/− versus WT yolk sac, calculated as a percentage of the total live cell population plus or minus SEM (n ≥ 3). Statistical analysis of significance was determined by Student t test, with a confidence interval of 95% (P ≤ .05).
Further analyses revealed a less severe effect of RA deficiency on non-SP cells. We observed a modest increase in the total number of non-SP cells in Raldh2−/− mutants compared with WT (4.5% vs 6.8%, P = .005; Figure 4A,B). As expected from our previous observations17 we found an increase in the endothelial cell fraction in E9.5 Raldh2−/− mutants compared with WT (1.7% vs 1.3%, P = .17; Figure 4C), and a trend toward an increase in committed hematopoietic cells (0.4% vs 0.7%, P = .06; Figure 4C). Together these data demonstrate that loss of RA signaling impairs development of hemogenic endothelium, resulting in loss of multipotent hematopoetic progenitor cell production.
RA signaling has been associated with cell survival in other model systems. Therefore, we examined the apoptotic index of Raldh2−/−-derived hemogenic endothelium relative to that of WT to determine whether an increased rate of death could explain their decreased numbers in vivo. To do so, we sorted SP cells from E9.5 WT and Raldh2−/− yolk sacs, and analyzed annexin V expression by FACS. Annexin V was detected on 57.5% plus or minus 18.0% of SP cells in Raldh2−/− mutants, compared with 27.1% plus or minus 19.0% in WT (P = .11). Together with previous studies demonstrating normal levels of apoptosis in the Raldh2−/− yolk sac mesoderm compared with WT at E8.5,17 these data indicate that increased cell death is not responsible for the loss of SP cells observed in E9.5 Raldh2−/− yolk sac.
Transcription of genes associated with hematopoiesis is decreased in the absence of RA signaling
Having correlated embryonic RA deficiency with decreased production of hemogenic endothelium and multipotent hematopoietic progenitors, we aimed to determine whether genes known to regulate definitive hematopoiesis were dysregulated in Raldh2−/− mutants. To quantify relative transcriptional expression of genes known to play a role in hematopoiesis in WT and RA-deficient mutants, we performed quantitative RT-PCR on RNA isolated from E8.5 and E9.5 WT and Raldh2−/− yolk sac tissues. Although hematopoietic genes expressed during early vasculogenesis showed normal expression in Raldh2−/− mutants before the time of hemogenic endothelial cell formation (E8.0; Figure 2D), all hematopoietic markers analyzed showed decreased expression in Raldh2−/− mutants at E8.5 (data not shown), and were significantly down-regulated at E9.5 compared with WT littermates (Figure 5A). Importantly, endothelial cell–specific genes were normally expressed in Raldh2−/− mutants (data not shown) as previously demonstrated,17 consistent with a role for RA signaling in the commitment of vascular endothelial cells to hemogenic fates, rather than in endothelial cell formation per se.

RA signaling regulates hematopoietic potential and function of hemogenic endothelium during murine embryogenesis. (A) qPCR analysis of hematoendothelial gene expression in E9.5 Raldh2−/− versus WT littermate yolk sacs. All data points were calculated as gene expression relative to endogenous β-actin expression (n = 3). (B) qPCR analysis of hematoendothelial gene expression in FACS-isolated subpopulations of E9.5 Raldh2−/− and WT littermate yolk sac. All data points were calculated as gene expression relative to endogenous β-actin expression (n = 3). (C) Quantitative profile of hematopoietic potential of E9.5 Raldh2−/− versus WT yolk sac. Data were calculated as the number of individual colonies generated per 1000 viable seeded cells plus or minus SEM (n ≥ 3).
RA signaling regulates hematopoietic potential and function of hemogenic endothelium during murine embryogenesis. (A) qPCR analysis of hematoendothelial gene expression in E9.5 Raldh2−/− versus WT littermate yolk sacs. All data points were calculated as gene expression relative to endogenous β-actin expression (n = 3). (B) qPCR analysis of hematoendothelial gene expression in FACS-isolated subpopulations of E9.5 Raldh2−/− and WT littermate yolk sac. All data points were calculated as gene expression relative to endogenous β-actin expression (n = 3). (C) Quantitative profile of hematopoietic potential of E9.5 Raldh2−/− versus WT yolk sac. Data were calculated as the number of individual colonies generated per 1000 viable seeded cells plus or minus SEM (n ≥ 3).
Although it was not technically possible to analyze gene expression profiles of Raldh2−/− versus WT hemogenic endothelium directly due to impaired formation in mutants, we clarified the relative contribution of hemogenic endothelium and multipotent hematopoietic progenitor cells to yolk sac expression of hematopoietic associated genes by performing qPCR on isolated subpopulations of the E9.5 WT yolk sac (Figure 5B). As expected, expression of transcription factors such as GATA-1, Lmo2, SCL/Tal-1, and Runx-1, were enriched in hemogenic endothelium (Flk1+CD45− SP) and/or multipotent hematopoietic progenitor cells (Flk1−CD45+ SP) relative to nonhemogenic endothelium (Flk1+CD45− non-SP). Consistent with its role in the transitional phenotype from endothelial cell to hematopoietic progenitor,26,27 CD41 was also shown to be highly enriched within the hemogenic endothelial cell population. Decreased expression of these critical hematopoietic genes in the E9.5 Raldh2−/− yolk sac is therefore consistent with the loss of hemogenic endothelial cell development and multipotent hematopoietic progenitor production observed in these animals.
Multilineage hematopoietic potential of hemogenic endothelium is impaired in the absence of RA signaling
We next investigated whether RA signaling regulates the hematopoietic potential of the few hemogenic endothelial cells that do form in Raldh2−/− mutants. Digested E9.5 WT and Raldh2−/− yolk sac tissues were fractionated into total SP and non-SP cells, cultured in MethoCult GF M3434 for up to 14 days and quantified by number and lineage type. Gross morphologic assessment of cultures revealed a significant decrease in overall hematopoietic colony-forming activity in Raldh2−/− yolk sacs compared with that of WT littermates. Hematopoietic colony-forming ability was enriched in WT yolk sac SP cell cultures, and only SP cells formed multilineage hematopoietic colonies (CFU-GEMM). Raldh2−/− SP cells yielded only erythroid (BFU-E) colonies, failing to produce either multilineage or myeloid (CFU-GM) colonies (Figure 5C).
We performed similar analyses of the hemogenic endothelium (Flk1+c-Kit+CD45− SP), and multipotent hematopoietic progenitor cells (Flk1−c-Kit+CD45+ SP) by subfractionating the SP cells isolated from E9.5 WT and Raldh2−/− yolk sacs: cultures of WT hemogenic endothelium exhibited enrichment of erythroid colony-forming ability, and similar myeloid and multilineage potential, compared with whole yolk sac SP (Figure 5C). As expected, the WT hematopoietic progenitor population was enriched for BFU-E, CFU-GM and CFU-GEMM colony-forming ability compared with whole SP. In contrast, RA-deficient hemogenic endothelium and multipotent hematopoietic progenitors failed to exhibit any hematopoietic colony-forming ability. These data reveal a critical and previously unrecognized role for RA signaling in regulating the potential of definitive hematopoietic progenitors, and the hemogenic endothelium from which they derive.
Restoration of RA signaling in vivo rescues differentiation of hemogenic endothelium
We have previously demonstrated that the vascular defects of Raldh2−/− embryos can be rescued by providing exogenous RA to mutant embryos via maternal diet supplementation.17 Here, we examined whether we could similarly rescue the observed defects in hemogenic endothelial cell formation and differentiation in RA-deficient mutants during the critical period of development when these processes are occurring (E7.5-9.5). Pregnant Raldh2+/− females were fed powdered chow supplemented with 100 μg/g all trans-retinoic acid (ATRA) from E7.5 until sacrifice at E9.5. WT embryos from RA-fed litters exhibited no teratogenic effects,16,17 and all Raldh2−/− embryos of RA-fed dams exhibited normal morphology upon dissection at E9.5. As expected, mutant embryos of control dams continued to display typical vascular defects of Raldh2-deficient embryos.
Hoechst profiling of RA-fed Raldh2−/− and WT littermate yolk sac cells revealed statistically significant rescue of the SP compartment in RA-fed mutants compared with WT (0.6% vs 0.6%, P = .27; Figure 6A,B). Both the hemogenic endothelial and hematopoietic progenitor subfractions of the SP were restored to WT levels (0.1% vs 0.1%, P = .23 and 0.1% vs 0.1%, P = .43, respectively; Figure 6C). The non-SP compartment of RA-fed mutants was also normalized, with vascular endothelial and committed hematopoietic cell populations comparable to WT levels (4.8% vs 4.7%, P = .41; Figure 6C).

Restoration of RA signaling in RA-deficient embryos rescues specification of hemogenic endothelium. (A) Phenotypic analysis of hemogenic specification in RA-treated E9.5 WT, and (B) E9.5 Raldh2−/− littermate yolk sac. (C) Quantitative analysis of the SP compartment in RA-treated E9.5 Raldh2−/− versus WT littermate yolk sac. All quantitative data were calculated as a percentage of the total live cell population plus or minus SEM (n ≥ 3). Statistical analysis of significance was determined by Student t test, with a confidence interval of 95% (P ≤ .05).
Restoration of RA signaling in RA-deficient embryos rescues specification of hemogenic endothelium. (A) Phenotypic analysis of hemogenic specification in RA-treated E9.5 WT, and (B) E9.5 Raldh2−/− littermate yolk sac. (C) Quantitative analysis of the SP compartment in RA-treated E9.5 Raldh2−/− versus WT littermate yolk sac. All quantitative data were calculated as a percentage of the total live cell population plus or minus SEM (n ≥ 3). Statistical analysis of significance was determined by Student t test, with a confidence interval of 95% (P ≤ .05).
Restoration of RA signaling restores hematopoietic gene expression
To determine whether restoration of RA signaling could rescue hematopoietic gene expression in Raldh2−/− mutants, we isolated RNA from the yolk sacs of RA-fed WT and Raldh2−/− littermate embryos, and performed qPCR for hematopoietic factors that we previously found to be down-regulated in response to RA deficiency. Expression of all genes was restored to at least 80% of WT levels (Figure 7A). These data confirm a functional role for RA in regulating expression of hematopoietic genes presumed to be involved in the specification and differentiation of hemogenic endothelium in vivo.

Restoration of RA signaling in RA-deficient embryos rescues hematopoietic potential and function of hemogenic endothelium. (A) qPCR analysis of endogenous hematoendothelial gene expression in RA-treated E9.5 Raldh2−/− versus WT littermate yolk sacs. All data were calculated as gene expression relative to endogenous β-actin expression of the tissue (n = 3). (B) Hematopoietic potential of RA-treated E9.5 Raldh2−/− mutant versus WT littermate yolk sac in vitro. All data were calculated as the number of individual colonies generated per 1000 viable seeded cells plus or minus SEM (n ≥ 3).
Restoration of RA signaling in RA-deficient embryos rescues hematopoietic potential and function of hemogenic endothelium. (A) qPCR analysis of endogenous hematoendothelial gene expression in RA-treated E9.5 Raldh2−/− versus WT littermate yolk sacs. All data were calculated as gene expression relative to endogenous β-actin expression of the tissue (n = 3). (B) Hematopoietic potential of RA-treated E9.5 Raldh2−/− mutant versus WT littermate yolk sac in vitro. All data were calculated as the number of individual colonies generated per 1000 viable seeded cells plus or minus SEM (n ≥ 3).
Restoration of RA signaling restores hematopoietic potential of hemogenic endothelium
To test whether restoration of RA signaling could rescue multipotent hematopoietic potential in Raldh2−/− mutants, we again isolated distinct cell populations from RA-fed E9.5 WT and Raldh2−/− littermate yolk sacs, and examined their function in hematopoietic colony-forming assays. Raldh2−/− SP cells with restored RA signaling demonstrated erythroid (BFU-E) and myeloid (CFU-GM) colony-forming potential similar to that of WT (Figure 7B), despite producing no multilineage (CFU-GEMM) colonies. When the SP was fractionated to isolate Flk1+c-Kit+CD45− hemogenic endothelium however, we found recovery of multilineage hematopoietic potential in RA-fed mutants to ∼66% of WT levels. In turn, we observed complete restoration of hematopoietic function in the Flk1−c-Kit+CD45+ multipotent hematopoietic progenitor population.
Discussion
Our understanding of the molecular pathways that govern the parallel development of blood and blood vessels is still in its infancy, and the molecular signals that direct the formation of hemogenic endothelium from unspecialized (primordial) vascular endothelium are completely unknown. In these studies, we have built upon previous work in which we isolated and phenotypically characterized hemogenic endothelial cells from the murine embryo and yolk sac.15 Herein, we demonstrate on a clonal level that yolk sac Flk1+c-Kit+CD45− SP cells represent a specialized subset of endothelial cells with multilineage hematopoietic potential (hemogenic endothelium).
We found that hemogenic endothelium represents approximately 10% of the total yolk sac SP, which itself constitutes approximately 0.5% to 1% of the total yolk sac cell population; thus, hemogenic endothelial cells constitute approximately 0.05% to 0.1% of yolk sac cells. This is consistent with observations in the adult bone marrow, in which the SP represents approximately 0.1% of the whole bone marrow cells and is highly enriched for adult hematopoietic stem cell activity.21 Our colony-forming data demonstrate enrichment of multilineage hematopoietic progenitor activity within the hemogenic endothelial cell fraction of the yolk sac SP. We therefore speculate that hemogenic endothelium functions to generate multilineage hematopoietic progenitors before establishment of the bone marrow niche, and herein begin to define the signaling pathway required for its formation and function in vivo.
Given the known importance of RA signaling for vasculogenesis16,17 and adult hematopoiesis,28,29 we hypothesized that RA signaling may play a role in the formation and function of hemogenic endothelium. Other studies have implicated RA in the regulation of developmental hematopoiesis,18,19,30 and although no specific cellular role for RA in this process has previously been identified, RA receptors are specifically expressed by endothelial cells at the initiation of definitive hematopoiesis.17 Our finding that Flk1+c-Kit+CD45− (SP) cells express RARα1/2 during this period of hematopoietic development suggests that RA signaling may regulate the formation and/or function of hemogenic endothelial cells in vivo.
To determine the role of RA signaling in developmental hematopoiesis, we examined RA deficient embryos for specific defects in primitive and definitive hematopoiesis. We found that the formation of primitive erythroblasts, primordial endothelium and vascular channels, and initiation of plasma flow was not impaired in Raldh2−/− embryos, indicating that RA signaling is not required for primitive hematopoiesis. We also found that development of Flk1+CD31+ SP cells, the immediate precursors to hemogenic endothelial cells that contribute to definitive hematopoiesis, formed normally in Raldh2−/− mutants. Differing levels of hemogenic endothelium observed in more mature WT and Raldh2−/− yolk sacs were therefore not the result of differing capacities to produce hemogenic endothelial cell precursors. These data are consistent with previous studies in the vitamin A–deficient (VAD) quail embryo that report normal development of the intraembryonic vasculature and yolk sac blood islands in the absence of retinoid signaling.31 No defects in primitive hematopoiesis have been described for any of the RAR32-34 or RXR receptor knockouts,35 providing further support for the idea that retinoid signaling is not essential for the earliest phase of hematopoietic development, but may instead modulate definitive hematopoiesis via the regulation of hemogenic endothelial cell development.
In addition to molecular signaling pathways that control endothelial cell phenotype and behavior during vasculogenesis, hemodynamic forces are also known to play an important role in this process. The developmentally timed emergence of hemogenic endothelium is coincident with the onset of systemic blood circulation, which alters mechanical forces within the vascular plexus and triggers vascular remodeling and endothelial cell specialization.6 Genomic studies have identified genes that are differentially regulated in endothelial cells by hemodynamic forces,36-38 which are known to directly impact the commitment of endothelial cells toward arterial and venous fates.39,40 Thus, it is possible that mechanical forces, as well as soluble signals, similarly impact the specialization of hemogenic endothelium from primordial endothelium in the vascular plexus. The fact that we found hemodynamic forces to be normal in RA-deficient mutants suggests that the observed defects in the development of hemogenic endothelium were not caused by global differences in mechanotransduction, but rather mediated by disrupted RA receptor-mediated signaling.
A molecular hierarchy of factors downstream of RA signaling that regulate the development of hemogenic endothelium has yet to be defined. However, our studies demonstrate that, in the absence of RA signaling, several transcription factors with known roles in hematopoietic development including GATA-1/2,41,42 SCL/Tal1,43,44 Lmo2,45 and Runx146 are down-regulated at the time of hemogenic endothelial cell formation and onset of definitive hematopoiesis. Embryos lacking any one of these transcription factors exhibit the same phenotype: formation of primordial endothelium, but impaired mature blood cell production.
Collectively, our findings suggest that RA signaling may modulate the expression of genes required for developmental hematopoiesis in the subset of endothelial cells that acquire hemogenic potential. Dissection of the signaling hierarchy downstream of RA that regulates the formation and function of hemogenic endothelium, although complex and beyond the scope of this current study, is needed to gain insight into the mechanism(s) of specialization of endothelial cells, and control of definitive hematopoiesis. Such information may also be relevant to the control of hematopoiesis in adult systems, and may prove useful for the optimization of clinical therapies for hematopoietic and/or vascular disorders.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by National Institutes of Health (Bethesda, MD) grants R01 EB005173 and P20 EB007076, and American Heart Association (Dallas, TX) EIA grant 0440054 AN (K.K.H.).
National Institutes of Health
Authorship
Contribution: L.C.G. and K.K.H. conceptualized and designed the research and authored the manuscript; L.C.G. performed all experiments and associated data analysis, with the exception of the FRAP experiments, which were designed and performed by M.E.D. and J.L.L. All listed authors contributed to revision of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Karen K. Hirschi, PhD, Center for Cell & Gene Therapy, Baylor College of Medicine, One Baylor Plaza, Room N1020, Houston, TX 77030; e-mail: khirschi@bcm.tmc.edu.

