

Abstract
Loss of neurofibromin or interferon consensus sequence binding protein (Icsbp) leads to a myeloproliferative disorder. Transcription of NF1 is directly controlled by ICSBP. It has been postulated that loss of NF1 expression resulting from loss of transcriptional activation by ICSBP contributes to human hematologic malignancies. To investigate the functional cooperation of these 2 proteins, we have established Icsbp-deficient mice with Nf1 haploinsufficiency. We here demonstrate that loss of Icsbp and Nf1 haploinsufficiency synergize to induce a forced myeloproliferation in Icsbp-deficient mice because of an expansion of a mature myeloid progenitor cell. Furthermore, Nf1 haploinsufficiency and loss of Icsbp contribute synergistically to progression of the myeloproliferative disorder toward transplantable leukemias. Leukemias are characterized by distinct phenotypes, which correlate with progressive genetic abnormalities. Loss of Nf1 heterozygosity is not mandatory for disease progression, but its occurrence with other genetic abnormalities indicates progressive genetic alterations in a defined subset of leukemias. These data show that loss of the 2 tumor suppressor genes Nf1 and Icsbp synergize in the induction of leukemias.
Introduction
Neurofibromatosis type I, or von Recklinghausen disease, is an autosomal dominant disorder with a prevalence of approximately 1:3000 worldwide. Carriers of the mutations develop benign neurofibromas and café-au-lait spots and are predisposed to neuronal tumors and to juvenile myeloid neoplasms, which include juvenile myelomonocytic leukemia (JMML), myelodysplasia (MDS), and acute myeloid leukemia (AML). In approximately 50% to 60% of children affected developing myeloid leukemias, loss of heterozygosity (LOH) of the normal NF1 allele is observed. In other cases, a mutant NF1 allele causes truncation and functional disability of the protein, neurofibromin 1.1,2 The NF1 promoter is transcriptionally regulated by RUNX1, C/EPBα, and ETS2 and dominant negatively suppressed by the product of the t(8;21) translocation fusion product AML-ETO in AML.3 This indicates that suppression of NF1 expression may also contribute to the development of AML in adults
Interferon consensus sequence binding protein (Icsbp), synonymous to interferon regulator factor 8, has dual functions in immunity and myelopoiesis. Icsbp belongs to the family of proteins regulating the cellular response to interferons, therefore baptized interferon-regulatory factors. Consequently, targeted deletion of Icsbp leads to the development of an immune deficiency syndrome toward certain viruses,4 intracellular bacteria,5 and parasites.6-8 Furthermore, Icsbp regulates myelopoiesis. Loss of Icsbp in mice leads to a myeloproliferative disorder (MPD) characterized by accumulation and expansion of mature neutrophil granulocytes and myeloid progenitor cells.4,9 This defect has been localized to the committed granulocyte-monocyte-progenitor (GMP),10 but the molecular mechanisms underlying the aberrant myeloid development are not understood today. Loss of ICSBP cooperates with several other human oncogenes in the induction of myeloid leukemias, including the 2 most common mutations in human myeloid malignancies, BCR/ABL11-15 and AML-ETO,16 as well as NUP98-TOP1–induced leukemias.17 Although mutations of the ICSBP gene have not been identified yet, hypermethylation of putative promoter regions has been found in human leukemias.18 Furthermore, activating SHP-2 mutations detected in human AML inhibit ICSBP function because of dephosphorylation of ICSBP with consequent loss of transactivation of the NF1 promoter.19 This implicates the dysregulation of neurofibromin 1 through loss of ICSBP function in the development and progression of human leukemias.
Leukemias developing in mice with deficiency of either neurofibromin or Icsbp show significant similarities in phenotype.4,20,21 Complete loss of either neurofibromin or Icsbp causes an MPD, which converts with a relatively low frequency to acute, predominant myeloid, leukemias. BXH-2 mice frequently develop leukemias through retroviral insertional mutagenesis into susceptible loci, one of which is the Evi-2 locus that disrupts Nf1 expression.22,23 Furthermore, BXH-2 mice carry a nonconservative mutation (915 C to T) resulting in an arginine-to-cysteine substitution at position 294 within the Icsbp-ORF. This mutation does not affect Icsbp expression, but splenocytes carrying this mutation were unable to produce interleukin-12 and interferon-gamma.24 This recessive mutation behaves therefore as a loss-of-function mutation of Icsbp, cooperating potentially with retroviral mutagenesis in the induction of leukemias. This model has been successfully used to search for potential AML-cooperating alterations by proviral insertional mutagenesis.25,26 These observations, the high phenotypic similarities, and the fact that ICSBP directly regulates the NF1 promoter27,28 prompted us to investigate whether the combined loss of neurofibromin and Icsbp will synergize in the progression from chronic MPD toward acute leukemia.
Methods
Mice
Icsbp Nf1 mice were generated by breeding Icsbp C57BL/6 mice4 with Nf1+/− mice (129/sv × C57BL/6 background)20 for several generations. Mice were bred under pathogen-free conditions. Experiments were performed in accordance with the German animal protection law. For all experiments, age- and sex-matched littermates were used.
Mouse analysis
Blood counts were determined by an automatic blood counter (VetABC; SCIL, Viernheim, Germany). Blood smears were prepared, May-Gruenwald-Giemsa stained, and analyzed by a blinded investigator on a Leica DMRB Microscope (Leica, Wetzlar, Germany) at ×400 magnification. Photomicrographs were acquired using the SpotColorMosaic 14.2 Video Camera and SpotSoftware 4.1 (Diagnostics Instruments, Sterling Heights, MI). Bone marrow (BM) cells were isolated and erythrocytes were lysed in PharmLyse lysis buffer (BD Biosciences, Heidelberg, Germany). Cryopreserved leukemia specimens from BM and spleen were thawed and transplanted at a dose of 1 to 2 × 106 via the tail vein of nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice. Some mice were irradiated with 235 cGy before transplantation.
Cultivation of BM macrophages
BM cells were cultivated for 7 to 10 days in Dulbecco modified Eagle medium (PAA Laboratories, Coelbe, Germany), 10% fetal calf serum (FCS; Biochrom, Berlin, Germany), 5% horse serum (Biochrom), 2 mM glutamine (PAA Laboratories), 1 mM sodium pyruvate (PAA Laboratories), 0.05% β-mercaptoethanol (Invitrogen, Karlsruhe, Germany), 1% penicillin/streptomycin (PAA Laboratories) supplemented with 20% conditioned medium from L929 cells.
CFU assay
A total of 3.3 × 104 BM or spleen cells were plated into 1 mL methylcellulose medium (MethoCult M3134), supplemented with 30% FCS, 2 mM glutamine, 2% penicillin/streptomycin, 0.1 mM β-mercaptoethanol in replica (primary culture). Recombinant cytokines were used in the following combinations: recombinant murine granulocyte-macrophage colony-stimulating factor (rmGM-CSF; 5 U/mL), rmGM-CSF (5 U/mL) plus recombinant rat stem cell factor (rrSCF; 20 ng/mL), recombinant murine interleukin-3 (5 U/mL) plus rrSCF (20 ng/mL). After determining the colony number on day 7, 3.3 × 104 cells recovered from the primary culture were plated in replica (secondary culture) using the same cytokines. The colony number was determined on day 10, respectively, 15 to 18 days after plating. Primary ckit+ cells were stimulated with increasing GM-CSF concentrations (0.1, 0.5, 1, and 5 U/mL) plus or minus SCF (10 ng/mL). A total of 0.5 × 104 cells per methylcellulose plate were plated and colonies counted on day 7. All colonies were enumerated under a stereo microscope (GC 6; Leica).
Sorting of Lin− and c-kit+ hematopoietic progenitor cells
Lineage-negative (Lin−) hematopoietic progenitor cells from BM were purified with LS columns (Miltenyi Biotec, Bergisch Gladbach, Germany) after incubation with biotin-conjugated Gr-1, CD11b, Ter119, CD3e, and B220 (all BD Biosciences) followed by streptavidin-coated magnetic microbeads (Miltenyi Biotec). For sorting of myeloid committed ckit+ cells, BM cell suspensions were prepared and cells were stained with c-kit phycoerythrin (PE) together with a mixture of Gr-1 fluorescein isothiocyanate (FITC) and CD11b (FITC) antibodies (BD Biosciences), and sorted on an Aria (BD Biosciences) high-speed flow cytometer.
Fluorescence-activated cell sorter analysis
Cells were resuspended in fluorescence-activated cell sorter (FACS) buffer (2% FCS, 2 mM ethylenediaminetetraacetic acid, 0.1% NaN3 in phosphate-buffered saline) supplemented with different antibodies (Gr-1-PE, CD11b-allophycocyanin, B220-FITC, CD16/32-PE, CD34-FITC, c-kit–allophycocyanin, FLT3-PE, SCA1-FITC, SCA1-biotin, Il-7Ra–biotin, CD31-biotin; all BD Biosciences). If using a biotinylated first antibody, a second staining with fluorochrome-coupled streptavidin was performed as indicated. The stained cells were washed, resuspended in FACS buffer, and analyzed with the FACSCalibur using CellQuest software (BD Biosciences).
Polymerase chain reaction and RT–polymerase chain reaction
Genomic DNA and total RNA were extracted from primary BM and spleen cells, respectively. The RevertAid H Minus first-strand cDNA synthesis kit (MBI Fermentas, St Leon Rot, Germany) was used for cDNA synthesis. Polymerase chain reaction (PCR) was performed with 50 to 100 ng genomic DNA or cDNA, respectively (Figure S5A, available on the Blood website; see the Supplemental Materials link at the top of the online article).
Quantitative real-time PCR
The real-time PCR was performed with the LightCycler system in combination with the LightCycler FastStart DNA MasterPlus SYBR green-1 kit (Roche Diagnostics, Mannheim, Germany). cDNA (70-200 ng) was used as a template (Figure S5A). After each cycle, the products were denatured and the fluorescence drop measured at a product-specific melting temperature (84°C Nf1, 89°C; glyceraldehyde-3-phosphate dehydrogenase [GAPDH]). The relative Nf1 (% Nf1) amount in relation to the GAPDH amount was calculated as follows:
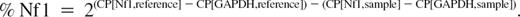
Histopathology
The organs (spleen, lymph nodes, and liver) were freshly fixed in 4% paraformaldehyde (in phosphate-buffered saline) and embedded in paraffin. Sections (5 μm thick) were stained with hematoxylin and eosin and analyzed by light microscopy (Olympus BX40 microscope equipped with a ColorviewII digital Camera and Software all Olympus Soft Imaging Solutions, Münster, Germany) by a board-certified pathologist (A.D.G.).
Results
Nf1 haploinsufficiency results in reduced Nf1 mRNA expression in different hematopoietic tissues
To investigate the cooperative loss of the tumor suppressor genes Nf1 and Icsbp for leukemia development, we crossed mice haploinsufficient for Nf1 into mice with a targeted deletion of Icsbp. We first quantified Nf1 wild-type mRNA transcripts using real-time PCR with primers specific for the wild-type allele. In our hands, neurofibromin expression could not be detected by Western blotting in primary murine hematopoietic cells. As composition of BM and spleen of wild-type and Icsbp-deficient mice was markedly different, we compared Nf1-mRNA expression in native BM and spleen separately in wild-type and Icsbp-deficient mice. Nf1 haploinsufficiency led to a significant reduction of more than 50% of Nf1 transcripts present in BM and spleen from Icsbp wild-type and Icsbp-deficient hematopoietic tissues (Figure 1). Probably because of the low Nf1 mRNA expression in Icsbp−/−Nf1+/− BM, individual measurements showed a high variation; therefore, this difference was not statistically different compared with Icsbp−/−Nf1+/+ BM.

Reduced Nf1 expression resulting from Nf1 heterozygosity. The amount of Nf1 transcripts was quantified by real-time PCR on cDNA obtained from total BM cells and spleen cell suspensions. Depicted is the mean relative expression ratio ± SD of Nf1 wild-type transcripts compared with expression of Gapdh mRNA (n = 5). Statistical significance (Mann-Whitney U test) is shown above the bars. (A) Icsbp+/+. (B) Icsbp−/−.
Reduced Nf1 expression resulting from Nf1 heterozygosity. The amount of Nf1 transcripts was quantified by real-time PCR on cDNA obtained from total BM cells and spleen cell suspensions. Depicted is the mean relative expression ratio ± SD of Nf1 wild-type transcripts compared with expression of Gapdh mRNA (n = 5). Statistical significance (Mann-Whitney U test) is shown above the bars. (A) Icsbp+/+. (B) Icsbp−/−.
We also compared expression of Nf1 wild-type transcripts in 5 independently generated BM macrophage lines (bone marrow macrophages [BMMs]) of all 4 genotypes. Nf1 expression was normalized for each individual experiment to Icsbp+/+Nf1+/+ (100%). Expression was significantly reduced in Icsbp+/+Nf1+/− (55.5% ± 9.4%; P = .016) and Icsbp−/−Nf1+/− (40.5% ± 24.1%; P = .008) but not significantly different in Icsbp−/−Nf1+/+ BMMs (58.5% ± 32.8%; P = .151). These findings confirm our results presented in Figure 1 as well as previous results from other investigators showing that expression of Nf1 depends on the numbers of functional alleles present.20 In contrast, for BMMs we could not confirm that Nf1 mRNA expression is regulated by Icsbp.19,27,28
Nf1 haploinsufficiency induces a forced but stable chronic MPD in Icsbp-deficient mice
Next, we investigated changes in the hematopoietic system induced through either individual loss of Icsbp or Nf1 haploinsufficiency or the combination of both mutations. Hematologic analysis of mice at the age of 2 to 4 months demonstrated a leukocytosis in Icsbp-deficient compared with wild-type mice as expected from previous analysis.4 Although complete loss of Nf1 expression leads to an MPD, this is not observed in Nf1-heterozygous mice,20,21 indicating that the MPD is caused by reduced Nf1 expression below a critical threshold. In Icsbp−/−Nf1+/− mice, we observed an aggravated leukocytosis because of an approximately 2-fold increase in neutrophil granulocytes in the peripheral blood (Figure 2A,B) but no absolute reduction of B220 lymphocytes (Figure S1A,B). The spleens of Icsbp−/−Nf1+/− were significantly larger than those of Icsbp−/−Nf1+/+ animals (Figure 2C), and lymph nodes showed a variable infiltration of neutrophil granulocytes (Figure 2D). Leukocytosis and splenomegaly, sensitive markers of myeloproliferation, remained constant up to 9 months, a time point before leukemia development (Figure 2A,C).

Enhanced myelopoiesis in Icsbp−/−Nf1+/− mice. (A) Highest total leukocyte counts from peripheral blood in Icsbp−/−Nf1+/− mice. Total leukocyte counts were determined using an automated hematologic cell counter from the peripheral blood (tail vein) from mice 2 to 4 months (young, n = 26) and 7 to 9 months of age (old, n = 16). Dots represent individual measurements; lines, medians; the level of significance comparing Icsbp−/−Nf1+/− and Icsbp−/−Nf1+/+ mice is shown above dots. Nonparametric test was used (Mann-Whitney U test) to calculate level of significance. (B) Highest neutrophil granulocyte counts in peripheral blood in Icsbp−/−Nf1+/− mice. Blood smears (n = 5) were prepared from peripheral blood, and total numbers of neutrophil granulocytes were determined blindly after May-Gruenwald-Giemsa staining. FACS analysis was performed after lysis of erythrocytes. Cells staining high with Gr-1 and CD11b were identified as neutrophil granulocytes. Absolute numbers of neutrophil granulocytes were calculated multiplying numbers of total leukocytes with the percentage of cells staining for Gr-1 and CD11b (n = 5). Dots represent individual measurements; lines, medians; the level of significance (Mann-Whitney U test) comparing Icsbp−/−Nf1+/− and Icsbp−/−Nf1+/+ mice is shown above dots. (C) Highest splenomegaly in Icsbp−/−Nf1+/− mice. Spleens and total body weights from mice 2 to 4 months (young, n = 25) and 7 to 9 months (old, n = 12) were determined using a precision balance. Dots represent individual measurements; lines, medians; the level of significance (Mann-Whitney U test) comparing Icsbp−/−Nf1+/− and Icsbp−/−Nf1+/+ mice is shown above dots. (D) Infiltration of neutrophil granulocytes into lymph nodes. Percentages of neutrophil granulocytes were determined using flow cytometry staining in BM, spleen, and lymph nodes from mice 4 months of age. Cells were pregated in a live gate and resolved for staining of Gr-1 and CD11b. Numbers above squares represent mean ± SD of n = 5 individual mice. Statistical significance (Mann-Whitney U test): *between Icsbp−/−Nf1+/+ and Icsbp+/+Nf1+/+; **between Icsbp−/−Nf1+/− and Icsbp+/+Nf1+/−.
Enhanced myelopoiesis in Icsbp−/−Nf1+/− mice. (A) Highest total leukocyte counts from peripheral blood in Icsbp−/−Nf1+/− mice. Total leukocyte counts were determined using an automated hematologic cell counter from the peripheral blood (tail vein) from mice 2 to 4 months (young, n = 26) and 7 to 9 months of age (old, n = 16). Dots represent individual measurements; lines, medians; the level of significance comparing Icsbp−/−Nf1+/− and Icsbp−/−Nf1+/+ mice is shown above dots. Nonparametric test was used (Mann-Whitney U test) to calculate level of significance. (B) Highest neutrophil granulocyte counts in peripheral blood in Icsbp−/−Nf1+/− mice. Blood smears (n = 5) were prepared from peripheral blood, and total numbers of neutrophil granulocytes were determined blindly after May-Gruenwald-Giemsa staining. FACS analysis was performed after lysis of erythrocytes. Cells staining high with Gr-1 and CD11b were identified as neutrophil granulocytes. Absolute numbers of neutrophil granulocytes were calculated multiplying numbers of total leukocytes with the percentage of cells staining for Gr-1 and CD11b (n = 5). Dots represent individual measurements; lines, medians; the level of significance (Mann-Whitney U test) comparing Icsbp−/−Nf1+/− and Icsbp−/−Nf1+/+ mice is shown above dots. (C) Highest splenomegaly in Icsbp−/−Nf1+/− mice. Spleens and total body weights from mice 2 to 4 months (young, n = 25) and 7 to 9 months (old, n = 12) were determined using a precision balance. Dots represent individual measurements; lines, medians; the level of significance (Mann-Whitney U test) comparing Icsbp−/−Nf1+/− and Icsbp−/−Nf1+/+ mice is shown above dots. (D) Infiltration of neutrophil granulocytes into lymph nodes. Percentages of neutrophil granulocytes were determined using flow cytometry staining in BM, spleen, and lymph nodes from mice 4 months of age. Cells were pregated in a live gate and resolved for staining of Gr-1 and CD11b. Numbers above squares represent mean ± SD of n = 5 individual mice. Statistical significance (Mann-Whitney U test): *between Icsbp−/−Nf1+/+ and Icsbp+/+Nf1+/+; **between Icsbp−/−Nf1+/− and Icsbp+/+Nf1+/−.
To define the deregulated progenitor pool that expands and is responsible for the enhanced MPD, we investigated defined progenitor pools (Figure S3A) by flow cytometry. We further examined Nf1 expression in the common myeloid progenitor (CMP) and GMP. Reverse-transcribed PCR (RT-PCR) of RNA isolated from wild-type and Icsbp-deficient CMP and GMP as well as analysis of global gene expression analysis of GMP performed previously in our laboratory revealed little to no detectable expression of Nf1 transcripts in either CMP or GMP (Figure S3B, Table S1, and Gene Expression Omnibus [NCBI GEO accession number GSM27216]29 ). In concert with these findings, the number of functional Nf1 alleles did not influence frequencies of progenitor populations in Icsbp-deficient mice (long-term hematopoietic stem cell [LT-HSC], Flt3−; Icsbp−/−Nf1+/−: 3.4% ± 0.9% vs Icsbp−/−Nf1+/+: 3.6% ± 0.9%, LMPP: 5.1% ± 1.6% vs 4.4% ± 1.3%; CMP: 1.5% ± 0.6% vs 1.0% ± 0.5%, and GMP: 21.7% ± 3.7% vs 22.0% ± 13.1%). Nf1 haploinsufficiency did also not affect frequencies of progenitor populations in wild-type mice (LT-HSC, Flt3−; Icsbp+/+Nf1+/−: 7.1% ± 1.3% vs Icsbp+/+Nf1+/+: 7.8% ± 1.2%, LMPP: 5.9% ± 1.0% vs 4.7% ± 1.9%; CMP: 2.9% ± 1.4% vs 3.1% ± 1.4%, and GMP: 10.3% ± 3.5% vs 9.9% ± 2.1%; Figure 3A). We also investigated Nf1 expression in lineage-committed myeloid progenitor cells. Again, Nf1 expression could not be detected in these cells (Figure S3B). As Nf1 is physiologically low or not expressed in those cells, this indicates that Nf1 haploinsufficiency does not alter myeloid differentiation up to the GMP or myeloid committed progenitor cells. In contrast, those progenitor compartments (LT-HSC, Flt3−; wild-type: 7.8% ± 1.2% vs Icsbp-def, 3.6% ± 0.9%, CMP 3.1% ± 1.4% vs 1.0% ± 0.5%, and GMP 9.9% ± 2.1% vs 22.0% ± 13.1%) were significantly different between wild-type and Icsbp-deficient mice (Figure 3A). Therefore, in contrast to Nf1, Icsbp affects myelopoiesis at an early myeloid committed progenitor stage.10

Distribution of progenitor cell populations. (A) Highest secondary CFU-GM formation in BM and spleen of Icsbp−/−Nf1+/− mice. BM cells from 2- to 3-month-old mice were lineage depleted and stained according to Kondo et al44 and Adolfsson et al,45 as described in “Fluorescence-activated cell sorter analysis” in “Methods.” Stem cell populations (Figure S3) were identified as follows: HSCs (Lin−, Sca1+, ckit+, Flt3−); LMPPs (Lin−, Sca1+, ckit+, Flt3+); CMPs (Lin−, Sca1−, ckit+, Flt3−, IL7Rα−, CD16/32intermediate, CD34intermediate); GMPs (Lin−, Sca1−, ckit+, Flt3−, IL7Rα −, CD16/32high, CD34high); megakaryocyte-erythrocyte progenitors (Lin− Sca1−, ckit+, Flt3−, IL7Rα−, CD16/32dim, CD34dim); and CLPs (Lin−, Sca1+, ckit+, IL7Rα+). Lineage-depleted cells were pregated as negative for lineage markers combined with a live gate. Percentages within gates represent the mean ± SD of Lin− cells of n = 3 individual mice of each genotype except for CLP (n = 2). Statistical significance (Mann-Whitney U test) *between Icsbp−/−Nf1+/+ and Icsbp+/+Nf1+/+ and **between Icsbp−/−Nf1+/− and Icsbp+/+Nf1+/−. (B,C) Increased secondary CFU-GM from BM and spleen. BM cells were grown in duplicates of 1 mL methylcellulose supplemented with combinations of rmGM-CSF and rrSCF or rrSCF and recombinant murine interleukin-3 (B). In addition, BM and spleen cells were grown in rmGM-CSF (C), as described in “CFU assay” in “Methods.” Cultures were performed in duplicates, incubated at 37°C with 7% CO2, and cultured for 8 to 10 days (Figure S2). At that time, colonies were counted, and all cells were isolated from methylcellulose and replated again into 1 mL methylcellulose in duplicates using the same cytokines as in the first CFU-GM assay. After 10 to 15 days, number of total colonies was scored again using a stereomicroscope. Dots represent numbers of secondary CFU-GM per total cells plated; and lines, medians. The level of significance (Mann-Whitney U test) comparing Icsbp−/−Nf1+/− and Icsbp−/−Nf1+/+ mice is shown above dots.
Distribution of progenitor cell populations. (A) Highest secondary CFU-GM formation in BM and spleen of Icsbp−/−Nf1+/− mice. BM cells from 2- to 3-month-old mice were lineage depleted and stained according to Kondo et al44 and Adolfsson et al,45 as described in “Fluorescence-activated cell sorter analysis” in “Methods.” Stem cell populations (Figure S3) were identified as follows: HSCs (Lin−, Sca1+, ckit+, Flt3−); LMPPs (Lin−, Sca1+, ckit+, Flt3+); CMPs (Lin−, Sca1−, ckit+, Flt3−, IL7Rα−, CD16/32intermediate, CD34intermediate); GMPs (Lin−, Sca1−, ckit+, Flt3−, IL7Rα −, CD16/32high, CD34high); megakaryocyte-erythrocyte progenitors (Lin− Sca1−, ckit+, Flt3−, IL7Rα−, CD16/32dim, CD34dim); and CLPs (Lin−, Sca1+, ckit+, IL7Rα+). Lineage-depleted cells were pregated as negative for lineage markers combined with a live gate. Percentages within gates represent the mean ± SD of Lin− cells of n = 3 individual mice of each genotype except for CLP (n = 2). Statistical significance (Mann-Whitney U test) *between Icsbp−/−Nf1+/+ and Icsbp+/+Nf1+/+ and **between Icsbp−/−Nf1+/− and Icsbp+/+Nf1+/−. (B,C) Increased secondary CFU-GM from BM and spleen. BM cells were grown in duplicates of 1 mL methylcellulose supplemented with combinations of rmGM-CSF and rrSCF or rrSCF and recombinant murine interleukin-3 (B). In addition, BM and spleen cells were grown in rmGM-CSF (C), as described in “CFU assay” in “Methods.” Cultures were performed in duplicates, incubated at 37°C with 7% CO2, and cultured for 8 to 10 days (Figure S2). At that time, colonies were counted, and all cells were isolated from methylcellulose and replated again into 1 mL methylcellulose in duplicates using the same cytokines as in the first CFU-GM assay. After 10 to 15 days, number of total colonies was scored again using a stereomicroscope. Dots represent numbers of secondary CFU-GM per total cells plated; and lines, medians. The level of significance (Mann-Whitney U test) comparing Icsbp−/−Nf1+/− and Icsbp−/−Nf1+/+ mice is shown above dots.
To test whether graded reduction of Nf1 expression because of Nf1 haploinsufficiency induced an increase in CFU formation in Icsbp-deficient cells, we compared colony formation of CFU-GM in response to GM-CSF, GM-CSF and SCF as well as IL-3 and SCF. In Icsbp-deficient mice, CFU-GM colony formation was significantly higher regardless of the Nf1 genotype after stimulation with GM-CSF (Figure S2A,B). CFU-GM formation in response to different cytokine combinations was not significantly different in either Icsbp wild-type Nf1+/+ or Nf1+/− mice as previously reported by others21,30 or Icsbp-deficient Nf1+/+ or Nf1+/− mice (Figure S2A,B). CFU-GM from Icsbp−/−Nf1+/− mice continued to grow after 8 days, whereas CFU-GM from Icsbp−/−Nf1+/+ mice did not. To substantiate this finding, we replated the cells from the primary CFU into secondary cultures at day 8 and incubated them for another 10 to 15 days. Here, secondary CFU-GM derived from Icsbp−/−Nf1+/− mice showed an approximately 3- to 4-fold higher clonogenicity than the ones derived from Icsbp−/−Nf1+/+ mice (Figure 3B,C). Primary clonogenicity of CFU-GM in response to GM-CSF from spleen was also not different (Figure S2A,B), whereas secondary colony formation was significantly increased in Icsbp−/−Nf1+/− mice (Figure 3C). These data indicate that the forced MPD in Icsbp−/−Nf1+/− mice is caused by an autochthonous capacity for prolonged proliferation or increased self-renewal of rather mature myeloid precursors from BM and spleen.
To investigate whether graded reduction of Nf1 expression in Icsbp−/−Nf1+/− leads to a measurable hyperactivation of the Ras- and/or AKT-kinase pathways or hypersensitivity toward different cytokines, we isolated c-kit-positive cells from the BM and stimulated them with GM-CSF and GM-CSF plus SCF. Form those sorted c-kit+ cells, we performed CFU assays with titered doses of those cytokines. Colony numbers were not different for either titered concentration of cytokines tested (Figure S2C). In parallel, we measured quantitatively the amount of phosphorylated ERK-1/2 and AKT via intracellular staining using flow cytometry in total BM. Although we were unable to detect Nf1 expression in sorted myeloid committed c-kit+ cells (Figure S3B), Nf1 expression could be readily detected in the BM of Icsbp-deficient mice (Figure 1). Therefore, potential biochemical consequences of reduced Nf1 expression should be detectable in those cells. As shown in Figure S2D, we could not detect a reproducible difference in either ERK-1/2 or AKT phosphorylation in Icsbp−/−Nf1+/− BM cells compared with Icsbp−/−Nf1+/+ BM cells.
Nf1 haploinsufficiency induces transplantable leukemias in Icsbp-deficient mice
Forced MPD in Icsbp−/−Nf1+/− mice led to an increased incidence of leukemias. Kaplan-Meier overall survival analysis showed that more than 50% of Icsbp−/−Nf1+/− mice died until the age of 11 months, whereas more than 80% of mice of the other 3 genotypes were still alive (Figure 4A). Analysis of moribund mice showed a high leukocyte count as an indication of leukemia in Icsbp−/−Nf1+/− mice, in some instances more than 200 000/μL peripheral blood. Median leukocyte counts, spleen, and liver weight, as an indication of infiltration with malignant cells, significantly increased in those mice (Figure 4B).

Overmortality of Icsbp−/−Nf1+/− mice resulting from leukemias. (A) Kaplan-Meier survival curves of Icsbp−/−Nf1+/− (●, n = 50); Icsbp−/−Nf1+/+ (○, n = 56); Icsbp+/+Nf1+/− (▲, n = 41); and Icsbp+/+Nf1+/+ (△, n = 32). (B) Comparison of total leukocyte counts, spleen weight normalized to body weight, and normalized liver weights of each individual diseased mouse. Dots represent individual measurements; and lines, medians. The level of significance (Mann-Whitney U test) comparing Icsbp−/−Nf1+/− and Icsbp−/−Nf1+/+ mice is shown above dots for Icsbp−/−Nf1+/− (n = 26); Icsbp−/−Nf1+/+ (n = 9); Icsbp+/+Nf1+/− (n = 8); and Icsbp+/+Nf1+/+ (n = 2). (C) Extensive enlargements of lymph nodes, liver, and spleen of Icsbp−/−Nf1+/− diseased mice. (D) Disease incidences and overall distribution of leukemias in all 4 genotypes. Mice were observed until the age of 15 months. Necropsy was performed on all mice either after the observation period or in case of clinical signs of disease. Some mice that died unexpectedly without disease manifestation (ie, dead without prior disease) could not be investigated. In some mice (moribund, no leukemia) cause of death could not be established, but a hematologic disease could be excluded. *Statistically significant difference (P < .001 by χ2 test) for the incidence of leukemia.
Overmortality of Icsbp−/−Nf1+/− mice resulting from leukemias. (A) Kaplan-Meier survival curves of Icsbp−/−Nf1+/− (●, n = 50); Icsbp−/−Nf1+/+ (○, n = 56); Icsbp+/+Nf1+/− (▲, n = 41); and Icsbp+/+Nf1+/+ (△, n = 32). (B) Comparison of total leukocyte counts, spleen weight normalized to body weight, and normalized liver weights of each individual diseased mouse. Dots represent individual measurements; and lines, medians. The level of significance (Mann-Whitney U test) comparing Icsbp−/−Nf1+/− and Icsbp−/−Nf1+/+ mice is shown above dots for Icsbp−/−Nf1+/− (n = 26); Icsbp−/−Nf1+/+ (n = 9); Icsbp+/+Nf1+/− (n = 8); and Icsbp+/+Nf1+/+ (n = 2). (C) Extensive enlargements of lymph nodes, liver, and spleen of Icsbp−/−Nf1+/− diseased mice. (D) Disease incidences and overall distribution of leukemias in all 4 genotypes. Mice were observed until the age of 15 months. Necropsy was performed on all mice either after the observation period or in case of clinical signs of disease. Some mice that died unexpectedly without disease manifestation (ie, dead without prior disease) could not be investigated. In some mice (moribund, no leukemia) cause of death could not be established, but a hematologic disease could be excluded. *Statistically significant difference (P < .001 by χ2 test) for the incidence of leukemia.
All mice were followed until the age of 15 to 19 months, a time that is close to the maximal life span of approximately 80% of mice in our facility. At this time point, a necropsy was performed on each animal. Massive hepatosplenomegaly and lymphadenopathy and organ infiltration with malignant leukemic cells were typically found in almost all diseased Icsbp−/−Nf1+/− mice (Figure 4C; Table 1). The number of mice found dead without prior disease manifestation, which therefore could not be investigated because of rapid organ autolysis, was similar in all 4 genotypes (Figure 4D). In an overall analysis, leukemia could be identified as the cause of death in 23 of 50 Icsbp−/−Nf1+/− mice. In contrast, only 4 of 56 of Icsbp−/−Nf1+/+ mice and 1 of 41 Icsbp+/+Nf1+/− mice developed leukemia (Figure 4D). None of the wild-type mice developed leukemia. This demonstrates that Nf1 haploinsufficiency induces progression from stable myeloproliferation toward frank leukemia in mice deficient for Icsbp.
Leukemias were classified according to established phenotypic criteria.30 In particular, MPD-like leukemias were characterized by extensive expansion of mature myeloid cells with percentages of blast cells in BM less than 20%. In contrast to mice with chronic MPD, mice with leukemia rapidly died of the disease with extensive with organ infiltration (Figure 4C). Acute leukemias were defined according to a consensus protocol31 by a blast percentage of more than 20% and either identification of myeloid or lymphoid markers as of myeloid (CD11b+, GR1+), lymphoid (B220+), or biphenotypic (CD11b+, B220+) acute leukemias (Figure S4); 47.8% of leukemias in Icsbp−/−Nf1+/− mice were found to be of MPD-like and 26.1% of myelomonocytic phenotype demonstrating that most of the leukemias that developed were of myeloid origin. MPD-like leukemias, in contrast to other leukemias, showed a drastic prolonged proliferative capacity or self-renewal. The number of second culture CFU-GM was at least 35 times higher than the number of CFUs from mice without leukemia and 10 times higher than the number of CFUs from myelomonocytic leukemias (not shown). The remaining leukemias were either of a lymphatic phenotype (13.0%), often with coexpression of the myeloid marker CD11b (biphenotypic), or displayed an undifferentiated phenotype (13.0%). Acute blastic leukemias, compared with MPD-like leukemias showed higher peripheral total leukocyte counts and in most cases an extensive infiltration of liver, spleen, and lymph nodes (Table 1). Representative examples for each leukemic phenotype with cytologic and immunophenotype, as well as histopathologic features of the affected organs, are shown in Figure 5.

Icsbp−/−Nf1+/− mice develop different types of leukemias; myeloid leukemias are predominant. Leukemias were classified according to diagnostic criteria established by Kogan et al.30 (A) Flow cytometry staining of leukemias: hematologic organs stained with the myeloid (Gr-1, CD11b) and a B-lymphoid marker (B220) showed differences in maturation and cell composition of the different forms of leukemia in comparison to mice without leukemias. MPD-like leukemias showed highest expression of myeloid and low expression of lymphoid markers in BM and spleen, whereas lymphatic leukemias showed little residual myelopoiesis in BM (n = 10, MPD-like myeloid leukemia; n = 5, myelomonocytic leukemia; n = 1, lymphatic leukemia: n = 5, preleukemic mice). (B) Cytomorphology of leukemias: myelomonocytic leukemias displayed a high percentage of myeloblasts with residual myeloid maturation, whereas lymphatic leukemias showed a high percentage of blast cells with little residual myeloid maturation. Mice without disease manifestation were characterized by high neutrophil granulocyte maturation as typical for Icsbp−/− mice (g indicates neutrophil granulocyte; l, lymphocyte; and b, blast). Pictures were taken with an Olympus BX41 microscope, equipped with a 40× UPlanFLN 0.75 objective with eyepiece UIS2 WHN 10×, in air. Slides were stained with hematoxylin and eosin. Electronic pictures were taken with a ColorView II version 2.0 camera (SIS, Münster, Germany). Images were acquired with analySIS docu, version 5.0 (SIS) and used without further processing. (C) Histology of tumor infiltrates. Disease-specific massive organ infiltration in spleen, submandibulary lymph nodes with extensive extramedullary hematopoiesis, and liver with mature and blastic (i-iii) in MPD-like leukemia or predominantly myeloblastic cells in myelomonocytic leukemia (iv-ix) was observed. Lymphatic leukemia showed infiltration with lymphocytes without differentiation pattern and large blasts (x-xii). In several instances, eosinophil crystalloid depositions (Charcot-Leyden crystals) were observed (ii). Organs from Icsbp−/−Nf1+/− mice without disease manifestation showed little infiltration with granulocytes and blasts (xiii-xv). Organs were stained with hematoxylin and eosin. Original magnification ×400. Pictures were taken with a Leica DMRB microscope, equipped with a 100× NPlan 1.25 objective with eyepiece magnification LPlan 10×, in oil. Slides were stained with May-Gruenwald. Electronic pictures were taken with an Insight camera 4.0 (Visitron Diagnostic Instruments, Sterling Heights, MI). Images were acquired and processed with SpotSoftware 4.1. (Visitron Diagnostic Instruments).
Icsbp−/−Nf1+/− mice develop different types of leukemias; myeloid leukemias are predominant. Leukemias were classified according to diagnostic criteria established by Kogan et al.30 (A) Flow cytometry staining of leukemias: hematologic organs stained with the myeloid (Gr-1, CD11b) and a B-lymphoid marker (B220) showed differences in maturation and cell composition of the different forms of leukemia in comparison to mice without leukemias. MPD-like leukemias showed highest expression of myeloid and low expression of lymphoid markers in BM and spleen, whereas lymphatic leukemias showed little residual myelopoiesis in BM (n = 10, MPD-like myeloid leukemia; n = 5, myelomonocytic leukemia; n = 1, lymphatic leukemia: n = 5, preleukemic mice). (B) Cytomorphology of leukemias: myelomonocytic leukemias displayed a high percentage of myeloblasts with residual myeloid maturation, whereas lymphatic leukemias showed a high percentage of blast cells with little residual myeloid maturation. Mice without disease manifestation were characterized by high neutrophil granulocyte maturation as typical for Icsbp−/− mice (g indicates neutrophil granulocyte; l, lymphocyte; and b, blast). Pictures were taken with an Olympus BX41 microscope, equipped with a 40× UPlanFLN 0.75 objective with eyepiece UIS2 WHN 10×, in air. Slides were stained with hematoxylin and eosin. Electronic pictures were taken with a ColorView II version 2.0 camera (SIS, Münster, Germany). Images were acquired with analySIS docu, version 5.0 (SIS) and used without further processing. (C) Histology of tumor infiltrates. Disease-specific massive organ infiltration in spleen, submandibulary lymph nodes with extensive extramedullary hematopoiesis, and liver with mature and blastic (i-iii) in MPD-like leukemia or predominantly myeloblastic cells in myelomonocytic leukemia (iv-ix) was observed. Lymphatic leukemia showed infiltration with lymphocytes without differentiation pattern and large blasts (x-xii). In several instances, eosinophil crystalloid depositions (Charcot-Leyden crystals) were observed (ii). Organs from Icsbp−/−Nf1+/− mice without disease manifestation showed little infiltration with granulocytes and blasts (xiii-xv). Organs were stained with hematoxylin and eosin. Original magnification ×400. Pictures were taken with a Leica DMRB microscope, equipped with a 100× NPlan 1.25 objective with eyepiece magnification LPlan 10×, in oil. Slides were stained with May-Gruenwald. Electronic pictures were taken with an Insight camera 4.0 (Visitron Diagnostic Instruments, Sterling Heights, MI). Images were acquired and processed with SpotSoftware 4.1. (Visitron Diagnostic Instruments).
To define the leukemic potential of the different leukemias as well as to investigate autochthonous growth in a host where Icsbp expression is otherwise normal, cryopreserved BM or spleen specimens of different phenotypes of leukemias were transplanted into NOD/SCID mice, as our mice were not congenic to an inbred mouse strain. Of all mice transplanted, 57.1% developed leukemia with a mean survival of 38.2 days. All mice (4 of 4) transplanted with a lymphatic leukemia, 80% (4 of 5) mice transplanted with a myelomonocytic leukemia, 75% (3 of 4) mice with a biphenotypic leukemia, and 44.4% (12 of 27) of all MPD-like leukemias died because of the transplanted leukemia. In all cases, transplanted leukemias faithfully replicated the cellular phenotype as well as the presence or absence of an LOH of the parental leukemia (Figure 6D). These findings indicated that leukemias arising in Icsbp-deficient mice are cell intrinsic and independent of the Icsbp-deficient environment. Furthermore, all different subtypes of leukemias were transplantable.

Disease incidences and LOH in established leukemias of Icsbp−/−Nf1+/− mice. (A) Allelic imbalance of Nf1 wt-allele expression in established leukemias. RT-PCR with allele specific primers was performed on cDNA-derived from BM of mice with established leukemias to investigate imbalanced expression of wild-type and mutated allele. The top band shows an amplification of a 511-bp fragment from the Nf1-wt allele transcript and the bottom band a 340-bp fragment of the Nf1-neomycin hybrid transcript. Actin expression was used as control. X denotes absence of wild-type band; (X), reduced wild-type band. Control lanes 8 to 12: mice without leukemia. (B) No allelic imbalance is observed in mice without disease manifestation. RT-PCR with allele specific primers was used to show balanced expression of wild-type and mutated Nf1 allele in young (3 months) and old (8-9 months) mice without disease manifestation. (C) Allelic imbalance of Nf1-wt allele expression in leukemias is the result of LOH. PCR with allele specific primers was performed on genomic DNA isolated from BM of diseased mice. In all but one case where an imbalanced allelic expression was observed, the wild-type band was lost or reduced in intensity. X indicates absence of wild-type band; (X), reduced wild-type band. (D) Replication of LOH in transplanted NOD/SCID mice. PCR with allele-specific primers was performed on genomic DNA of established leukemias isolated from BM of diseased NOD/SCID mice transplanted with 6 different leukemia entities from Icsbp−/−Nf1+/− mice. In all cases where LOH was observed in the parental leukemia, this was also observed in the diseased NOD/SCID mice. As a comparison are shown amplicons from genomic DNA from untreated NOD/SCID mice and from normal Nf1+/+ or Nf1+/− mice.
Disease incidences and LOH in established leukemias of Icsbp−/−Nf1+/− mice. (A) Allelic imbalance of Nf1 wt-allele expression in established leukemias. RT-PCR with allele specific primers was performed on cDNA-derived from BM of mice with established leukemias to investigate imbalanced expression of wild-type and mutated allele. The top band shows an amplification of a 511-bp fragment from the Nf1-wt allele transcript and the bottom band a 340-bp fragment of the Nf1-neomycin hybrid transcript. Actin expression was used as control. X denotes absence of wild-type band; (X), reduced wild-type band. Control lanes 8 to 12: mice without leukemia. (B) No allelic imbalance is observed in mice without disease manifestation. RT-PCR with allele specific primers was used to show balanced expression of wild-type and mutated Nf1 allele in young (3 months) and old (8-9 months) mice without disease manifestation. (C) Allelic imbalance of Nf1-wt allele expression in leukemias is the result of LOH. PCR with allele specific primers was performed on genomic DNA isolated from BM of diseased mice. In all but one case where an imbalanced allelic expression was observed, the wild-type band was lost or reduced in intensity. X indicates absence of wild-type band; (X), reduced wild-type band. (D) Replication of LOH in transplanted NOD/SCID mice. PCR with allele-specific primers was performed on genomic DNA of established leukemias isolated from BM of diseased NOD/SCID mice transplanted with 6 different leukemia entities from Icsbp−/−Nf1+/− mice. In all cases where LOH was observed in the parental leukemia, this was also observed in the diseased NOD/SCID mice. As a comparison are shown amplicons from genomic DNA from untreated NOD/SCID mice and from normal Nf1+/+ or Nf1+/− mice.
Phenotypes of leukemias correlate with genetic abnormalities and LOH
Because leukemias are often associated with distinct genetic abnormalities, we performed spectral karyotyping (SKY) analysis of metaphases isolated from BM cells and array comparative genomic hybridization (CGH) to detect genetic abnormalities. In all cases analyzed, lymphatic or biphenotypic leukemias showed different chromosomal gains by array CGH (Table 1; Figure S5B). SKY could not be performed, as the malignant lymphatic cells could not be propagated in cell culture to obtain metaphases. In contrast, in myeloid leukemias, genetic abnormalities were infrequent. No aberrations were detected in MPD-like leukemias by either SKY or array CGH (P = .029, Fisher exact test; Table 1). In agreement, in myelomonocytic leukemias, except for a gain of an X chromosome in one case, no additional abnormalities could be observed. These data suggest that acute leukemias with a switch from a myeloid to a lymphoid phenotype might be characterized by progressive genomic alterations.
JMML associated with neurofibromatosis 1 is often characterized by an LOH of the NF1 wild-type allele.1 In addition, mouse models suggest that Nf1 deficiency is mandatory to develop leukemias.32,33 To test whether this is also the case in our mouse model, we investigated the loss of wild-type allele expression resulting from LOH in established leukemias. Before the development of leukemias, only 1 of 12 mice of the older age group showed an imbalanced allelic expression of Nf1 (Figure 6B). In addition, most MPD-like leukemias did not show detectable LOH (Table 1; Figure 6A,C). Only in the 2 oldest mice, a complete or partial loss of the wild-type Nf1 mRNA transcript could be observed because of a LOH. In contrast, all myelomonocytic, lymphatic, or biphenotypic leukemias showed an LOH (P = .007, Fisher exact test; Table 1; Figure 6A,C). Furthermore, the presence of genomic aberrations as detected by array CGH was significantly associated with the presence of LOH (P = .048, Fisher exact test; Table 1).
Thus, in our experimental model: (1) an LOH is not mandatory for disease progression from chronic MPD toward lethal leukemia; and (2) the phenotypic classification of leukemias arising in Icsbp-deficient mice correlates with the presence or absence of global genetic abnormalities, including LOH.
Discussion
Nf1 haploinsufficiency has no obvious effect on Ras and AKT signaling in Icsbp−/− cells
Haploinsufficiency or deficiency of Nf1 in mice leads in several, but not all, cases to increased Ras activation in hematopoietic cells with the consequence of reduced apoptosis and increased proliferation.32,34-37 This results in a hypersensitivity toward several hematopoietic cytokines, notably GM-CSF and SCF.21,36,38,39 Furthermore, ICSBP directly regulates the NF1 promoter and reduced Nf1 expression has been implicated to contribute to the MPD observed in Icsbp-deficient mice.19,27,28 In our work, we demonstrate that Nf1 haploinsufficiency led to reduction of Nf1 expression in different hematopoietic tissues. Pairwise comparison between wild-type and Icsbp-deficient mice showed a reduction of Nf1 expression in the BM and spleen because of Nf1 haploinsufficiency. The different composition of the BM and spleen between Icsbp-deficient and wild-type mice precluded a direct comparison of all 4 genotypes. A direct comparison of Nf1 expression in BMMs, however, revealed an approximately 50% reduction of Nf1 expression in either Icsbp+/+Nf1+/− or Icsbp−/−Nf1+/+ cells; albeit in the latter, Nf1 expression was more variable and the difference did not reach statistical significance. This may have been different if a distinct progenitor cell population had been purified (see next paragraph). Therefore, haploinsufficiency induces a consistent reduction of Nf1 expression. Loss of Icsbp transactivation may induce a rather variable reduction of Nf1 expression in different hematopoietic cell types.
Nf1 haploinsufficiency induced neither hypersensitivity against GM-CSF or SCF, nor could we detect a hyperactivation of the RAS or AKT pathway in cells where we could readily detect significant differences in Nf1 expression. This may be the result of the cells that we selected for biochemical analysis. Because it is of obvious, pivotal interest to identify the cell population that is expanded in Icsbp−/−Nf1+/− mice causing the forced myeloproliferation, we performed an extensive phenotypical and Nf1 expression analysis of the myeloid progenitor hierarchy. Unfortunately, we could not determine the target population. A small, so far undefined, subgroup of progenitor cells might be responsible for the observed myeloid expansion. It is conceivable that biochemical alterations in RAS or AKT signaling could become apparent because of reduced Nf1 expression if assays were performed in such a progenitor cell population. Nevertheless, we were unable to detect differences related to haploinsufficiency of Nf1 in the size of well-defined progenitor populations, which can be prospectively isolated in either wild-type or Icsbp-deficient mice. In BM cells where we could readily detect significant differences in Nf1 expression, we were unable to detect differences in pERK or pAKT phosphorylation. Graded reduction of Nf1 expression to the extent that we observed may either not be sufficient to significantly affect signaling via the RAS and AKT pathways in Icsbp-deficient Nf1-haploinsufficient cells or may be operative only in a so for unidentified myeloid progenitor population.
Myeloid expansion through combination of Icsbp deficiency and Nf1 haploinsufficiency
Nf1 haploinsufficiency induced a forced MPD in the hematopoietic system of Icsbp-deficient mice. Reduced Nf1 expression did not affect early stages of hematopoietic development as phenotypically defined progenitor cells (GMP, megakaryocyte-erythrocyte progenitor, and CMP), and different hematopoietic stem cell compartments were not altered in either Icsbp-deficient or wild-type mice with a heterozygous deletion of Nf1. This is consistent with the fact that we could not detect Nf1 expression in neither GMP nor CMP. In contrast, loss of Icsbp led to a significant increase in numbers of GMP and a reduction of FLT3− HSC. This indicates that loss of transactivation of the Nf1 promoter resulting from the lack of Icsbp is not causally involved in the initiation of the MPD at an early progenitor cell. Nf1 haploinsufficiency becomes phenotypically evident in rather mature cell compartments. This is substantiated by the fact that (1) there was a consistent increase in the percentage and absolute numbers of mature neutrophil granulocytes in Icsbp−/−Nf1+/− mice compared with Icsbp−/−Nf1+/+ and (2) the myeloid compartment was globally significantly enlarged as evidenced by the increase in spleen size. The enormous spleen of Icsbp−/− mice sequestered a significant part of all myeloid cells. Splenomegaly is a clinical hallmark of several myeloproliferative diseases, including JMML.40 Finally, there was an increase of CFU-GM because of a capacity for prolonged proliferative capacity or self-renewal of those cells. The prolonged proliferation or self-renewal is most probably the cause for the enhanced granulocytosis and myeloproliferation observed in Icsbp−/−Nf1+/− mice. Enhanced neutrophil granulocytosis, splenomegaly, and prolonged proliferation or self-renewal were only observed in double-mutant mice, indicating that loss of Icsbp and Nf1 haploinsufficiency act synergistically on the expansion of myelopoiesis. Nf1 expression was readily detected in the BM as well as in the spleen, which was composed primarily of lymphocytes and mature cells of late-stage granulocyte precursors and neutrophils. Therefore, Nf1 expression increases with maturation, and loss of function of Nf1 becomes evident in rather mature progenitor and differentiated hematopoietic cells.
Cooperative leukemogenesis
Despite the lower expression of Nf1 in Icsbp−/−Nf1+/+ and Icsbp+/+Nf1+/− mice compared with wild-type mice, respectively, these mice rather seldom developed leukemias. The observed reduction of Nf1 expression was obviously not sufficient to induce leukemias. These findings suggest the existence of a threshold of myeloproliferative activity that needs to be overcome before leukemias develop. Oncogenes (or loss of tumor suppressor genes) cooperating in the same pathway may increase the myeloproliferative activity over this critical threshold. Alternatively or additionally, Nf1 haploinsufficiency may promote stochastically the complete loss of Nf1 expression because of either LOH or mutations in the coding sequence of Nf1. In our mice, LOH was not mandatory for disease progression and observed almost exclusively in acute leukemia subtypes with an aberrant lymphatic phenotype, especially the ones characterized by additional genetic alterations (in 4 of 5 cases analyzed). In our opinion, it is doubtful that a disabling coding sequence mutation of Nf1 rather than a LOH represents the leukemia-initiating event exclusively in MPD-like leukemias. The consequence of LOH and/or mutations in the coding sequence of Nf1 as leukemia-initiating events would lead to an identical cellular phenotype and should therefore not segregate with distinct phenotypes of leukemias. On the other hand, loss of Nf1 in the course of leukemia progression may contribute to the aggressive and invasive phenotype observed in acute leukemias with LOH in our study. As for MPD-like leukemia, the majority of cells that have still preserved the wild-type Nf1 allele might mask a LOH in a subclone. Yet by the definition of a tumor suppressor gene and the supporting literature, those neurofibromin-deficient cells should be clonally selected during the subclinical disease progression when transplanted into NOD/SCID mice and produce a detectable “de novo” LOH in diseased mice. In none of the transplanted MPD-like leukemias were we able to find such a de novo LOH, arguing against the hypothesis of a masked LOH in MPD-like leukemias.
Progression of leukemias
The long latency without obvious changes in the hematopoietic system and the sudden development of leukemia indicate that accumulation of additional mutations might be necessary for leukemia development. The nature of those mutations may then determine the phenotype of leukemias. It is further conceivable that those mutations affect transcription factors known to regulate Nf1 expression as, for example, Pu.127 or Runx1, C/ebpα, and Ets2.3 Whereas most leukemias were of myeloid phenotype, MPD-like leukemia phenotype, and myelomonocytic leukemia, the remaining leukemias demonstrated a lymphatic phenotype, in some cases with additional expression of the monocytic marker CD11b. Aggressivity of leukemias, as indicated by total leukocyte counts, spleen, and liver infiltration, progressed from MPD-like leukemias toward lymphatic leukemias (Table 1).
Acute leukemias were characterized by an increase in genetic alterations. LOH and other genetic abnormalities were significantly more frequent in leukemias deviating phenotypically from the initial chronic MPD observed in Icsbp−/−Nf1+/+ or Icsbp−/−Nf1+/− mice. After completion of primary analysis, we observed one case of a lymphatic leukemia without LOH (Figure 6). LOH was not observed in mice developing MPD-like leukemia younger than 12 months. Indeed, only in 2 mice developing MPD-like leukemia at the age of 15 months was an LOH observed. Whereas LOH is obviously not mandatory for MPD-like leukemia development, LOH during disease progression might select for a more aggressive and immature leukemic phenotype. Alternatively, the Nf1 wild-type allele might get lost as a consequence of increasing global genetic fragility. Indeed, several lines of evidence indicate that the presence of LOH depends on the nature of genotoxic stress and the availability of subsequent DNA repair mechanisms. LOH was reported to be associated with leukemia arising in Nf1 heterozygous mice treated with etoposide, but not with alkylating agents where LOH were equally frequent in mice with or without leukemia41 or radiation42 where, in contrast to solid tumors, no LOH was observed in leukemias. Furthermore, no LOH has been observed in all leukemias developing in an Nf1-haploinsufficient tumor model crossed into mice with a defect in the DNA mismatch repair enzyme Mlh1.43 Conditional deletion of a floxed Nf1 allele in Icsbp−/−Nf1f1/− should allow to mimic LOH and to study the consequences of the complete loss of Nf1 for the development of leukemias in mice.
In conclusion, we demonstrate in a novel in vivo mouse model, how mutations affecting the expression of those 2 proteins can cooperate in the induction or progression of hematopoietic neoplasms. Loss of Icsbp and Nf1 heterozygosity led to an expansion of a mature hematopoietic progenitor population, causing a forced MPD followed by a high incidence of leukemias. This observation may help to explain cooperative leukemogenesis in JMML as well as adult AML in which NF1 expression is compromised by the presence of oncogenic fusion proteins. AML-ETO has been shown to reduce the expression of NF13 and cooperates with loss of Icsbp in the induction of blastic transformation.16 Finally, the high incidence of leukemias in Icsbp−/−Nf1+/− mice, in concert with the long latency of disease development, will allow to prospectively study the nature of additional acquired genetic changes necessary for disease progression and the multistep process of leukemogenesis.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Claudia Pallasch and Bianca Verret for excellent animal care, Tanja Hinrichsen for performing the SKY analyses, Gregorcz Terszowski for sorting of GMP, Dido Lenze for performing the Affymetrix Array analysis, I. Horak and T. Moritz for critically reading the manuscript and stimulating discussions, and Carol Stocking for kindly providing us with NOD/SCID mice.
This work was supported by the Deutsche Forschungsgemeinschaft (CA 306/1-1; D.C.) and in part by the Deutsche José Carreras Stiftung e.V. (DJCLS R 05/22; L.B.). The Leibniz-Institut für Molekulare Pharmakologie is supported by the City of Berlin, Germany and the Bundesministerium für Bildung und Forschung, Berlin, Germany.
Authorship
Contribution: J.K., C.R., S.S., O.K., and L.B. designed and performed research and analyzed data; and A.D.G., B.S., and D.C. conceived and designed research, analyzed data, and wrote and edited the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Dirk Carstanjen, Leibniz-Institut für Molekulare Pharmakologie, Krahmer Strasse 6, D-12207 Berlin, Germany; e-mail: carstanjen@fmp-berlin.de.


{kind=link}
{kind=link}