Abstract
Platelet response to activation varies widely between individuals but shows interindividual consistency and strong heritability. The genetic basis of this variation has not been properly explored. We therefore systematically measured the effect on function of sequence variation in 97 candidate genes in the collagen and adenosine-diphosphate (ADP) signaling pathways. Resequencing of the genes in 48 European DNA samples nearly doubled the number of known single nucleotide polymorphisms (SNPs) and informed the selection of 1327 SNPs for genotyping in 500 healthy Northern European subjects with known platelet responses to collagen-related peptide (CRP-XL) and ADP. This identified 17 novel associations with platelet function (P < .005) accounting for approximately 46% of the variation in response. Further investigations with platelets of known genotype explored the mechanisms behind some of the associations. SNPs in PEAR1 associated with increased platelet response to CRP-XL and increased PEAR1 protein expression after platelet degranulation. The minor allele of a 3′ untranslated region (UTR) SNP (rs2769668) in VAV3 was associated with higher protein expression (P = .03) and increased P-selectin exposure after ADP activation (P = .004). Furthermore the minor allele of the intronic SNP rs17786144 in ITPR1 modified Ca2+ levels after activation with ADP (P < .004). These data provide novel insights into key hubs within platelet signaling networks.
Introduction
Platelets play a pivotal role in thrombus formation during normal hemostatic responses to injury and atherothrombotic disease such as myocardial infarction (MI) and stroke. They are rapidly activated at sites of vascular injury or plaque rupture by a range of physiologic agonists, including collagen, present in the vessel wall and found in elevated levels in atherosclerotic plaques,1 and adenosine diphosphate (ADP), released from activated platelets and damaged cells.
Several recent large-scale studies have unequivocally confirmed the long held notion that the response of platelets to all agonists is highly variable within the population.2-8 Interestingly the level of responsiveness within an individual is remarkably consistent over time, regardless of agonist, or function outcome measure.2-4,8-10 This, coupled with evidence from studies in siblings,11 twins,12 and in families with a history of premature coronary artery disease (CAD),13 suggests a high level of heritability of platelet response. It is therefore of interest to define the genetic loci that regulate this continuous quantitative trait (QT). Furthermore, the discovery of quantitative trait loci (QTLs) that regulate platelet function will help to clarify the relative importance of proteins in the complex networks that regulate platelet function.
To date, most investigations of such QTLs have focused on platelet surface receptors; particularly those responsible for fibrinogen-mediated platelet aggregation via the integrin αIIbβ3,14-18 or adhesion to collagen via α2β1,19,20 or von Willebrand factor via GPIbα,21-24 or the platelet signaling receptors for most of the physiologic agonists.2-4,10,25,26 The robustness of the observed genetic associations has been limited for many studies by small cohort sizes and, with a few exceptions, by inadequate selection of single nucleotide polymorphisms (tagSNPs) to ascertain underlying sequence variation.27 So, while the reported associations for C807T (rs1126643) in ITGA2, which encodes the α2 integrin,20 the GP6 haplotype,25 and the Kozac sequence in GP1BA,23,28 have been replicated, others have been less reproducible.29,30 Surprisingly nearly all genes encoding proteins in signaling cascades downstream of the cell surface receptors have remained unexplored.
We postulated that, as is the case for other highly heritable QTs such as height,31 platelet response would be controlled by a large number of genes each exerting a small effect. Furthermore, we reasoned that sequence variation in some QTLs would be pathway-specific, while others, particularly those encoding proteins operational downstream of the release of intracellular Ca2+, would be independent of the agonist used. It follows from these assumptions that studies that aim to identify platelet function QTLs require large cohorts of individuals, accurate and reproducible ascertainment of the functional trait, and genotyping for a large set of SNPs to enable sequence haplotypes to be inferred at an r2 value exceeding 0.8.
The present study takes the first steps in an effort to systematically unravel the complex relationship between genetic variation and platelet function. The first step was to establish the Platelet Function Cohort (PFC) of 500 healthy individuals. The characteristics of the PFC have been reported elsewhere8 but in short, platelet function was determined by measuring fibrinogen bound to activated αIIbβ3 (a prerequisite for aggregation) and P-selectin expression (a marker of platelet degranulation) after activation with either the glycoprotein VI (GPVI)–specific collagen mimetic, collagen-related peptide (CRP-XL), or ADP. These agonists were selected because they perform a major role in platelet activation after atherosclerotic plaque rupture and importantly represent 2 distinct signaling cascades, an early one via the FcRγ/ITAM signaling pathway (GPVI) and a later one which consolidates and amplifies platelet activation through 2 G protein–coupled receptors (GPCR), P2Y1 and P2Y12. The second step was the selection of 97 candidate genes based on a priori knowledge of the key proteins with known roles in either or both signaling cascades.32 The third step was to enhance information about sequence variation in the candidate genes in 48 reference DNA samples from the Center d'Etude du Polymorphism Humain (CEPH) European cohort. From the enriched dataset, SNPs were identified by a comprehensive tagging algorithm to test each gene for sequence variation at an r2 greater than or equal to 0.9, and all SNPs with consequences (nonsynonymous, alternative splice sites, and highly conserved noncoding regions [HCNCR]) were included. This resulted in 1327 SNPs that were genotyped in the 500 DNA samples from the PFC.
The results of the genotype-phenotype association study confirmed our earlier observation that the GP6 locus is a strong QTL for the platelet response to CRP-XL. In addition, we identified 14 novel QTLs with 17 independently associated SNPs, 12 in genes encoding intracellular proteins and 5 in genes encoding membrane receptors. Finally we performed follow-up studies on 3 genes, PEAR1, VAV3, and ITPR1, to define the mechanism by which sequence variation modifies platelet function.
Methods
Blood collection
A cohort of 500 healthy subjects of predominantly Northern European origin was recruited from the National Health Service Blood and Transplant blood donor clinic in Cambridge after gaining informed, written consent in accordance with the Declaration of Helsinki (for details of the cohort see Jones et al8 ). The study was approved by the Huntingdon Research Ethics Committee. Blood was drawn before donation, from the antecubital fossa vein, contralateral to the one used for routine donation, via a 21-gauge butterfly needle and a vacutainer tube (Becton Dickinson), using a standardized protocol designed to minimize artefactual platelet activation33 and was used within 10 minutes of donation. The first 3 mL blood were discarded, and subsequent samples were taken into 0.105 M sodium citrate.
Flow cytometric measurement of platelet activation
Platelet response to agonists was measured in 500 subjects by flow cytometry as described previously.34 Briefly, whole blood was incubated for 20 minutes with either 10−7 M ADP (Sigma), or 0.1 μg/mL CRP-XL (monomeric sequence GCI[GPO]10GCOG),34 and with either fluorescein isothiocyanate (FITC)–anti-fibrinogen (Dako Ltd) or phycoerythrin (PE)–anti-CD62P (Bristol Institute for Transfusion Science, Bristol, United Kingdom) in the presence of specific inhibitors of the main subsidiary activation pathways; namely 100 μM aspirin, 10 U/mL hirudin, and, for CRP-XL–stimulated samples, 4 U/mL apyrase (all from Sigma). Data were recorded as percentage of cells positive for the activation markers. The raw data were transformed to a logit scale and then fitted to a linear regression model, including confounding variables.8 The standardized residuals from the regression model were used in the genotype-phenotype association analysis throughout this paper.
Flow cytometric measurement of protein levels
PEAR1 levels were determined by resuspending washed platelets at 3 × 108/mL in N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES)–buffered saline (HBS) containing 5 mM glucose, 2 mM CaCl2, and 10 U/mL hirudin (HBS-GCH). Samples remained unstimulated or were stimulated with a combination of 1 μg/mL CRP-XL and 10−5 M thrombin receptor activating peptide (TRAP) for 45 minutes at 37°C to cause full degranulation (assessed by P-selectin expression; data not shown). They were subsequently incubated for a further 15 minutes with mouse polyclonal antisera raised against PEAR1 (see the supplemental methods, available on the Blood website; see the Supplemental Materials link at the top of the online article). Platelets were then washed, resuspended in HBS-GCH, incubated with FITC rabbit anti–mouse immunoglobulin G (IgG) at 37°C for 15 minutes, fixed using 0.2% formyl saline, and analyzed by flow cytometry.
To measure ITPR1 and VAV3, platelet-rich plasma (PRP) was fixed with 2% formyl saline, permeablized using BD Phosflow perm buffer III (BD Bioscience), washed, resuspended in HBS, and incubated with rabbit antibodies to ITPR1 (Abcam) or VAV3 (Millipore) for 30 minutes at 37°C. Platelets were then washed, resuspended in HBS, incubated for 30 minutes at 37°C, with PE-donkey-anti–rabbit IgG (Abcam), fixed using 0.2% formyl saline, and analyzed by flow cytometry. Negative controls for the ITPR1 and VAV3 antibodies were set using an appropriate isotype control. For PEAR1 preimmune sera from the same mouse was used.
GPVI expression was measured by the addition of the GPVI antibody HY101 to PRP for 30 minutes. The platelets were then washed and incubated with FITC rabbit anti–mouse IgG (Dako) for a further 30 minutes.
Flow cytometric analysis was performed using a Beckman-Coulter EPICS Profile XL or, for the measurement of ITPR1 and VAV3, a CyAn-ADP (Beckman-Coulter Ltd).
Calcium flux [Ca2+]i
Measurements were carried out using a Fluo-4 NW calcium assay kit (Molecular Probes). Briefly, an equal volume of HBS containing Fluo-4 NW dye and probenecid was added to PRP along with 100 μM aspirin, 10 U/mL hirudin, and, for the CRP-XL stimulated samples, 4 U/mL apyrase, and incubated at 37°C for 30 minutes. After incubation, platelets were stimulated with ADP (10−7, 10−6, 10−5 M) or CRP-XL (5 μg/mL), and [Ca2+]i was measured using a Fluoroskan Ascent plate reader (Thermo Labsystems) with excitation at 485 nm and emission measured at 538 nm.
Selection of candidate genes
Candidate genes were selected by prior knowledge of genes involved in the platelet signaling pathways downstream of GPVI and purinergic receptors, P2Y1 and P2Y12, which are stimulated by CRP-XL or ADP, respectively. Selection included genes coding for most of the relevant cell-surface agonist and adhesion receptors, and many genes encoding intracellular proteins with a well established function in either or both signaling pathways, for which there was absence of information on the relationship between sequence variation and cellular function (eg, VAV1, VAV3). Some genes (eg, PEAR1) were included because of new information emerging at the time of selection. The expression patterns of the candidate genes in blood cells were then investigated using data in the HaemAtlas.35
Exoseq
A validated pipeline for exon resequencing was used to identify sequence variants in genomic regions of interest. The exons and flanking intronic regions of all 97 candidate genes, plus the HCNCRs, which are enriched for SNPs regulating gene transcription,36 of 4 of the genes, (GP6, ITGA2, ITGB1, and CD109), were sequenced in 48 reference DNA samples from the CEPH European cohort. This resulted in an enrichment of SNPs that were not available in the reference SNP database, dbSNP, at the time of the release of our sequencing results, many of which showed a minor allele frequency (MAF) less than or equal to 0.05. The resequencing process involved the sequencing of polymerase chain reaction (PCR)–amplified genomic DNA of 48 persons from the CEPH pedigrees, 30 of which are shared with HapMap (Coriell Cell Repositories). Details of the process together with protocols are available in the supplementary methods and at http://www.sanger.ac.uk/humgen/exoseq/.
Golden Gate genotyping
SNP typing was performed on a custom Golden Gate array (Illumina) according to the manufacturer's protocol. In brief, 750 ng genomic DNA was PCR-amplified, then hybridized to a Sentrix Array Matrix (SAM; Illumina). The SAM was scanned using a BeadArray reader and the resulting intensity files were loaded into Illumina Beadstudio software, and manual cluster analysis was performed. QC analysis was performed, and only SNPs with cluster scores greater than 0.3 and call rates greater than 80%, were included in the output files. In addition, Hardy-Weinberg equilibrium (HWE) values less than 0.001 were examined, and samples not conforming were removed. Duplicate samples were included on each individual plate to assess genotyping error rates.
Statistical analysis
The association between each SNP and the 4 quantitative platelet response phenotypes (FA, FC, PA, and PC) was tested using an additive disease model (using PLINK, http://pngu.mgh.harvard.edu/purcell/plink). The regression coefficient and Wald test asymptotic P value are reported.
The linear models describing the combined effect of several SNPs on platelet activity were generated by stepwise regression using forward selection. The SNPs that could potentially contribute to the model were all those that associate with the platelet response phenotype with P less than .01, and these were added in order of descending significance. We present the proportion of variance explained by the SNPs in the final model.
For the analysis of [Ca2+]i curves, the peak value and log10 area under the curve (AUC) were fitted with a linear mixed model with fixed effects for genotype and dose, and random effect for individual. Time to peak (TTP) was modeled using Cox regression.
Results
The PFC
Four functional phenotypes were measured in the PFC of 500 healthy individuals, P-selectin expression in response to ADP (PA) and CRP-XL (PC) and fibrinogen binding in response to the same 2 agonists (FA and FC, respectively). As described previously, an intermediate dose of each agonist was used to ensure maximum discrimination of the differences in response between subjects, and pathway specificity was ensured by blocking the secondary agonists thrombin, thromboxane, and ADP in CRP-XL–stimulated samples.8 As illustrated in Figure 1, a wide variation in platelet response to stimulation existed within the population, and there was both concordance and divergence between the 4 phenotypes. Unsurprisingly, the greatest correlations observed were between QTs that share a common agonist, for example, PC and FC.

Platelet functional response phenotype in the platelet function cohort. Platelet response to ADP and CRP-XL was measured by flow cytometry in the 500 subjects in the platelet function cohort. Data are shown as the standardized residuals of logit transformed data, after adjustment for confounding variables, as described by Jones et al.8 Fibrinogen binding in response to CRP-XL (FC) is shown on the x-axis, with P-selectin expression in response to CRP-XL (PC) on the y-axis. P-selectin expression in response to ADP (PA) is shown on the z-axis, and fibrinogen binding in response to ADP (FA) is shown by color gradient from red (low response) to green (high response). Correlations (r2 values) are given for the linear models, comparing all 4 variables P < .00001 in all cases).
Platelet functional response phenotype in the platelet function cohort. Platelet response to ADP and CRP-XL was measured by flow cytometry in the 500 subjects in the platelet function cohort. Data are shown as the standardized residuals of logit transformed data, after adjustment for confounding variables, as described by Jones et al.8 Fibrinogen binding in response to CRP-XL (FC) is shown on the x-axis, with P-selectin expression in response to CRP-XL (PC) on the y-axis. P-selectin expression in response to ADP (PA) is shown on the z-axis, and fibrinogen binding in response to ADP (FA) is shown by color gradient from red (low response) to green (high response). Correlations (r2 values) are given for the linear models, comparing all 4 variables P < .00001 in all cases).
Expression of candidate genes in hematopoietic cells
Ninety-seven candidate genes were selected based on prior knowledge of their involvement in the platelet signaling pathways downstream of GPVI and the purinergic receptors, P2Y1 and P2Y12. Transcript levels were obtained from the HaemAtlas,35 and hierarchical clustering of the results from the 8 main blood cell elements indicates that their expression in megakaryocytes (MKs) is distinct from the 7 other blood cells (supplemental Figure 1), reflecting the importance of the proteins encoded by the candidate genes to MK and platelet biology.
Enrichment of SNPs and selection of SNPs for genotyping
At the time this experiment was designed, SNPs with MAFs less than or equal to 0.1 were known to be underrepresented in dbSNP. To capture variants with a MAF of 0.05, 0.03, and 0.01 with a 99%, 95%, and 87% chance, respectively, we sequenced 48 samples37 culminated in the identification of 2683 SNPs, of which 1068 (39.8%) were not present in dbSNP (build 126; supplemental Figure 2; the full dataset is available from www.bloodomics.org). Of all the SNPs identified, 788 (29%) had a MAF less than 0.03, of which 82% were novel, and 67 (2.5%) had a MAF less than 0.01, of which 92% were novel.
A comprehensive algorithm selected the optimal set of SNPs for genotyping (see the supplementary methods), resulting in a panel of 1327 SNPs (supplemental Table 1) that represented directly, or through tagging, 78% of the SNPs identified by sequencing and 95% of the polymorphic HapMap SNPs. The selected SNPs were mounted on a bespoke Illumina Golden Gate SNP genotyping array.
Identification of SNPs associated with platelet response
DNA from the 500 individuals in the PFC was genotyped for the selected 1327 SNPs (supplemental Table 1). After genotyping, an initial quality control, based on missingness and the HWE, was applied. Four DNA samples did not produce results of adequate quality and were removed, and 3 SNPs, 1 each in PLCB1, NFE2L2, and MADD, had highly significant evidence of deviation HWE. A cluster of SNPs in ITGB3 also showed deviation from HWE, the most significant P value being 2.4 × 10−5, which is due to a selection bias caused by the overrepresentation of ITGB3*002 homozygotes (rs5918, HPA-1b) in the PFC.38 None of the SNPs associating with the platelet responses showed significant deviation from HWE. To identify SNPs that were associated with platelet function, a P value less than or equal to .005 was applied to correct for multiple testing.
We have previously reported that GP6 genotype has a significant effect on platelet response to CRP-XL.8,25 This was confirmed, with 33 of the 42 GP6 SNPs showing an association with response to CRP-XL, but not ADP (P < .005, supplemental Table 2). The most significantly associated GP6 SNP, rs1613662, accounted for 38.6 and 27.1% of variation in PC and FC, respectively (P < .001, supplemental Table 2). rs1613662 is a nonsynonymous SNP that alters the residue 219 of GPVI from serine (major allele) to proline (minor allele), as previously reported by Watkins et al.27 Incorporation of rs1613662 genotype as a covariate in a linear model testing association with PC and FC demonstrated that none of the remaining GP6 SNPs showed further association (P > .01). Since a substantial proportion of response to CRP-XL is determined by variation in GPVI, both rs1613662 genotype and the GPVI protein expression level (because 10.8% of the variation in GPVI expression was explained by genotype, P < .001)8 were included as covariates in all subsequent linear models for testing association with FC and PC.
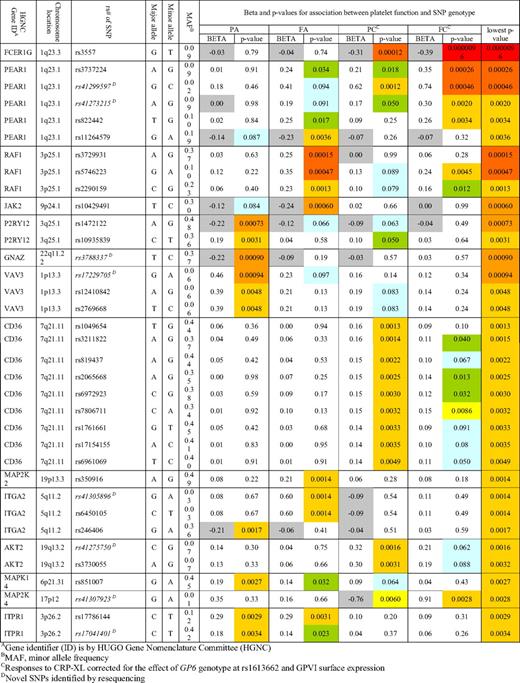
After accounting for the effect of GPVI, 35 SNPs, representing 14 genes, were associated with one or more of the platelet function measurements at a P value of less than .005 and a MAF of greater than 0.01 (Table 1, Figure 2, and supplemental Table 1). No SNPs were identified that affected all 4 measures of the platelet response. Four genes (CD36, FCER1G, MAP2K4, and PEAR1) showed association with both readouts in response to CRP-XL, and in addition, PEAR1 also showed an independent association signal with FA. One gene, AKT2, contained 2 SNPs that were associated only with PC. Specificity for the ADP signaling pathway was seen in 8 genes, 2 of which (ITGA2 and ITPR1) were linked to both readouts, while the rest contained SNPs whose effects were restricted to either fibrinogen binding (JAK2 and MAP2K2) or P-selectin expression (GNAZ, MAPK14, P2RY12, and VAV3). Finally a single gene, RAF-1, was linked to the level of fibrinogen binding in response to both agonists.

Venn diagram showing the associations between QTLs and platelet function. SNPs in 15 QTLs showed an association with one or more of the platelet function measurements at the P < .005 level and with a MAF > 0.01. The gene names of the QTLs are distributed among this Venn diagram according to the functional traits (PA, FA, PC, or FC) with which they are associated. Association with platelet functional measurement for each QTL, is shown by the colored squares that indicate the level of significance from blue (P < .10), green (P < .05), yellow (P < .01), gold (P < .005), light orange (P < .0005), dark orange (P < .001), and red (P < .0001).
Venn diagram showing the associations between QTLs and platelet function. SNPs in 15 QTLs showed an association with one or more of the platelet function measurements at the P < .005 level and with a MAF > 0.01. The gene names of the QTLs are distributed among this Venn diagram according to the functional traits (PA, FA, PC, or FC) with which they are associated. Association with platelet functional measurement for each QTL, is shown by the colored squares that indicate the level of significance from blue (P < .10), green (P < .05), yellow (P < .01), gold (P < .005), light orange (P < .0005), dark orange (P < .001), and red (P < .0001).
Additive effect of SNPs and proportion of variation explained
Further analysis, combining the effects of multiple SNPs, with a slightly relaxed P value of association less than or equal to .01 being accepted, showed that 38.4% of the total variation seen in the FC trait is determined by 10 SNPs (P < .001; Figure 3). Similarly combinations of SNPs accounted for 45.8% of variation in PC (P < .001), and 15.6 and 13.4%, respectively, of variation in FA (P < .001) and PA (P < .001; Figure 3).

Additive effect of SNPs on the platelet response. Each panel shows the additive association (r2) between SNPs and each platelet functional trait. To generate these models, all SNPs were added stepwise into a linear regression model in order of the significance of their individual associations (most significant first) and rejected if no longer significant (P > .01) when part of the combined model. All genotyped SNPs were entered into this model if they had an individual association with functional trait of P < .01.
Additive effect of SNPs on the platelet response. Each panel shows the additive association (r2) between SNPs and each platelet functional trait. To generate these models, all SNPs were added stepwise into a linear regression model in order of the significance of their individual associations (most significant first) and rejected if no longer significant (P > .01) when part of the combined model. All genotyped SNPs were entered into this model if they had an individual association with functional trait of P < .01.
SNPs within the same locus show significant associations with different functional measurements
There were several examples of SNPs in weak linkage disequilibrium (LD) in the same locus that showed independent associations with the platelet responses to different agonists. The most striking example of this was observed in PEAR1 (see below), but it was also observed for ITGA2, where the 2 rare SNPs, rs41305896 and rs6450105 (MAF = 0.030 for both), were associated with FA, while the common SNP, rs246406 (MAF = 0.359), showed association with PA but not with FA. We also identified SNPs within the same locus that showed independent associations for the same agonist but with opposing directionality. For example, 2 SNPs in P2RY12 showed an association with P-selectin expression in response to ADP (PA) but, while for 1 (rs1472122) the minor allele was associated with a higher response (P = .0007), the other (rs10935839) minor allele was associated with a lower response (P = .0031).
Confirmation and elucidation of function effects of SNPs
In this study, we have identified 14 novel QTLs for platelet responses, which illuminate aspects of signaling pathways. To confirm these effects and to explore the mechanism by which sequence variation alters platelet behavior, 3 genes (PEAR1, VAV3, and ITPR1) were selected for further study by recalling subjects from the PFC on the basis of known genotype.
Of the 14 SNPs tested in the PEAR1 locus, 5 showed association with platelet function. Four of these SNPs (rs3737224, rs41273215, rs41299597, and rs822442), which lie within a 4-kb region and are in strong LD, showed a strong association with FC and weaker association with PC (Figure 4A). Two of these introduced the nonconservative amino acid replacements S802C (rs41299597) and N848K (rs822442) in the cytoplasmic domain of PEAR1. Intriguingly a fifth PEAR1 SNP (rs11264579, MAF = 0.190) in the 5′ noncoding region showed a significant and independent association with FA (P = .0036), but no association with FC and PC. This SNP is in low LD with the other 4 (d′ = 0.60-0.91, r2 = 0.06-0.29; and see Figure 6A).
![Figure 4. Associations and LD structure for PEAR1. (A) Association of the 14 SNPs in the PEAR1 locus on chromosome 1q23.1, for each of the 4 functional platelet responses (reading from the top: PA, PC, FA, and FC). Arrows indicate the SNPs with the highest association in each analysis. The correlative triangles indicate the strength of LD between SNPs, as estimated from HapMap data. The colors vary from white (disequilibrium coefficient < 1; logarithm of the odds [LOD] score < 2) to blue (disequilibrium coefficient = 1; LOD score < 2) to pink (disequilibrium coefficient < 1; LOD score ≥ 2) to bright red (disequilibrium coefficient = 1; LOD score ≥ 2). The chromosomal position (in Mb) is based on National Center for Biotechnology Information (NCBI) and build 36 coordinates, showing Ensembl (version 49) gene structure. (B) Twenty-six donors recalled on the basis of combined genotype for rs3737224 and rs41299597 showed an increase in the expression of PEAR1 protein on the surface of platelets stimulated with both 1 μg/mL CRP-XL and 10−5 M TRAP. This increase was larger in subjects carrying the minor allele for both SNPs (P = .001). Data are shown as the mean and 75 and 95% ranges for each genotype.](/view-large/figure/7061634/zh89990938850004.jpeg)
Associations and LD structure for PEAR1. (A) Association of the 14 SNPs in the PEAR1 locus on chromosome 1q23.1, for each of the 4 functional platelet responses (reading from the top: PA, PC, FA, and FC). Arrows indicate the SNPs with the highest association in each analysis. The correlative triangles indicate the strength of LD between SNPs, as estimated from HapMap data. The colors vary from white (disequilibrium coefficient < 1; logarithm of the odds [LOD] score < 2) to blue (disequilibrium coefficient = 1; LOD score < 2) to pink (disequilibrium coefficient < 1; LOD score ≥ 2) to bright red (disequilibrium coefficient = 1; LOD score ≥ 2). The chromosomal position (in Mb) is based on National Center for Biotechnology Information (NCBI) and build 36 coordinates, showing Ensembl (version 49) gene structure. (B) Twenty-six donors recalled on the basis of combined genotype for rs3737224 and rs41299597 showed an increase in the expression of PEAR1 protein on the surface of platelets stimulated with both 1 μg/mL CRP-XL and 10−5 M TRAP. This increase was larger in subjects carrying the minor allele for both SNPs (P = .001). Data are shown as the mean and 75 and 95% ranges for each genotype.
Associations and LD structure for PEAR1. (A) Association of the 14 SNPs in the PEAR1 locus on chromosome 1q23.1, for each of the 4 functional platelet responses (reading from the top: PA, PC, FA, and FC). Arrows indicate the SNPs with the highest association in each analysis. The correlative triangles indicate the strength of LD between SNPs, as estimated from HapMap data. The colors vary from white (disequilibrium coefficient < 1; logarithm of the odds [LOD] score < 2) to blue (disequilibrium coefficient = 1; LOD score < 2) to pink (disequilibrium coefficient < 1; LOD score ≥ 2) to bright red (disequilibrium coefficient = 1; LOD score ≥ 2). The chromosomal position (in Mb) is based on National Center for Biotechnology Information (NCBI) and build 36 coordinates, showing Ensembl (version 49) gene structure. (B) Twenty-six donors recalled on the basis of combined genotype for rs3737224 and rs41299597 showed an increase in the expression of PEAR1 protein on the surface of platelets stimulated with both 1 μg/mL CRP-XL and 10−5 M TRAP. This increase was larger in subjects carrying the minor allele for both SNPs (P = .001). Data are shown as the mean and 75 and 95% ranges for each genotype.
Twenty-six subjects, comprising 4 groups were recalled for further investigation on the basis of their genotype for rs41299597 and rs3737224, the 2 SNPs with the lowest P value for association with PC and FC, respectively. Surface expression of PEAR1 was measured on washed platelets before and after degranulation in response to a combination of 1 μg/mL CRP-XL and 10−5 M TRAP. In resting (unstimulated) platelets, there was no significant difference in PEAR1 expression between individuals of the different genotypes. However, surface expression of PEAR1 increased after stimulation and was significantly greater in subjects carrying the minor allele of both SNPs (P = .001; Figure 4B), suggesting that the cytoplasmic cysteine may regulate the amount of intracellular PEAR1 that can traffic to the membrane upon stimulation.
Three of the 17 SNPs in VAV3 (rs17229705, rs12410842, and rs2769668) that are in strong LD showed an association with PA (Figure 5A). Seventeen subjects were recalled based on their genotype for rs17229705 (Figure 5A). Subjects who were heterozygous for this SNP had significantly increased abundance of VAV3 in their platelets compared with individuals who where homozygous for the major allele (Figure 5B-C). Because of the low MAF for all 3 SNPs, no PFC subjects homozygous for the minor allele were identified.

Associations and LD structure for VAV3. (A) Association of the 17 SNPs in the VAV3 locus on chromosome 1p13.3 for each of the 4 functional platelet responses (reading from the top; PA, PC, FA, and FC). Arrow indicates the SNP with the highest association in the PA response. LD structure is based on HapMap data (as described for Figure 4), and chromosomal position (in Mb) based on NCBI build 36 coordinates, showing Ensembl (version 49) gene structure. (B) Expression of VAV3 protein measured by flow cytometry in unstimulated platelets of subjects homozygous (GG) for the major allele of rs17229705 (solid line) or heterozygous (GA; dotted line). (C) Expression of VAV3 protein in platelets from 17 subjects who were either homozygous major (GG) or heterozygous (GA) for rs17229705.
Associations and LD structure for VAV3. (A) Association of the 17 SNPs in the VAV3 locus on chromosome 1p13.3 for each of the 4 functional platelet responses (reading from the top; PA, PC, FA, and FC). Arrow indicates the SNP with the highest association in the PA response. LD structure is based on HapMap data (as described for Figure 4), and chromosomal position (in Mb) based on NCBI build 36 coordinates, showing Ensembl (version 49) gene structure. (B) Expression of VAV3 protein measured by flow cytometry in unstimulated platelets of subjects homozygous (GG) for the major allele of rs17229705 (solid line) or heterozygous (GA; dotted line). (C) Expression of VAV3 protein in platelets from 17 subjects who were either homozygous major (GG) or heterozygous (GA) for rs17229705.
ITPR1 encodes the receptor for inositol triphosphate (IP3) and is central to calcium mobilization and hence the propagation of platelet intracellular signaling. One of the 61 SNPs tested in the ITPR1 locus, rs17786144 (MAF = 0.125, Table 1 and Figure 6A) showed a significant association with both PA and FA (P = .0029 and .003, respectively). Eighteen subjects were recalled on the basis of genotype for rs17786144, and platelet [Ca2+]i was measured in response to ADP and CRP-XL. The rs17786144 SNP showed a significant association with platelet [Ca2+]i in response to ADP (Figure 6B), but no association with the response to CRP-XL, which is in line with the original functional data in the 500 subjects in the PFC. In the response to ADP, the AUC was significantly increased with the minor allele (at ADP 10−7 M: CC = 20.4 ± 2.3, CT = 23.9 ± 4.0, TT = 25.6 ± 4.4; ADP 10−6 M: CC = 28.5 ± 2.6, CT = 29.6 ± 4.4, TT = 29.6 ± 5.4; ADP 10−5 M: CC = 22.7 ± 2.5, CT = 28.0 ± 6.5, TT = 29.9 ± 7.7; all units are arbitrary levels of fluorescence at 538 nm, P = .004). Similarly, the minor allele was associated with a delay in the time to peak (TTP) of 39% in the heterozygous compared with the major homozygous subjects (P = .013). This lengthening of TTP does not indicate a slower rate of [Ca2+]i, as there was no difference in maximum rate of flux between genotypes, but is indicative of a more sustained rise in intracellular calcium. The effect of ITPR1 genotype on platelet function was not due to measurable differences in protein levels (Figure 6C-D) suggesting that the intronic SNP rs17786144, modifies the platelet response to ADP by another, as yet unresolved, mechanism.

Associations and LD structure for ITPR1. (A) Association of the 61 SNPs in the ITPR1 locus on chromosome 3p26.1 for each of the 4 functional platelet responses (reading from the top; PA, PC, FA, and FC). Arrow indicates the SNP with the highest association in the PA response. LD structure is based on HapMap data (as described for Figure 4), and chromosomal position in Mb based on NCBI build 36 coordinates, showing Ensembl (version 49) gene structure. (B) Calcium flux (shown as area under the curve) in response to 3 concentrations of ADP (10−7, 10−6, 10−5 M) and 5 μg/mL CRP-XL. Data are shown as the mean ± for subjects homozygous for the major allele of rs17786144 (CC) (■; n = 10), heterozygous (CT) ( , n = 7), or homozygous for the minor allele (TT) (□, n = 4). (C) Calcium flux (shown as time to peak calcium) in response to 3 concentrations of ADP (10−7, 10−6, 10−5 M) and 5 μg/mL CRP-XL. Data are shown as the mean ± SD for subjects homozygous for the major allele or rs17786144 (CC) ■; n = 10), heterozygous (CT) (; n = 7), or homozygous for the minor allele (TT) (□; n = 4). (D) Example of flow cytometric histogram of expression of ITPR1 protein in platelets from subjects homozygous for the major allele of rs1786144 (solid line), heterozygous (dotted line), or homozygous for the minor allele (dashed line), illustrating the lack of effect of genotype on the level of expression. (E) Expression of ITPR1 protein, measured by flow cytometry, in platelets from 10 subjects homozygous for the major allele of rs1786144 (CC), 7 heterozygotes (CT), and 4 subjects homozygous for the minor allele (TT). (ns for all).
, n = 7), or homozygous for the minor allele (TT) (□, n = 4). (C) Calcium flux (shown as time to peak calcium) in response to 3 concentrations of ADP (10−7, 10−6, 10−5 M) and 5 μg/mL CRP-XL. Data are shown as the mean ± SD for subjects homozygous for the major allele or rs17786144 (CC) ■; n = 10), heterozygous (CT) (; n = 7), or homozygous for the minor allele (TT) (□; n = 4). (D) Example of flow cytometric histogram of expression of ITPR1 protein in platelets from subjects homozygous for the major allele of rs1786144 (solid line), heterozygous (dotted line), or homozygous for the minor allele (dashed line), illustrating the lack of effect of genotype on the level of expression. (E) Expression of ITPR1 protein, measured by flow cytometry, in platelets from 10 subjects homozygous for the major allele of rs1786144 (CC), 7 heterozygotes (CT), and 4 subjects homozygous for the minor allele (TT). (ns for all).
Associations and LD structure for ITPR1. (A) Association of the 61 SNPs in the ITPR1 locus on chromosome 3p26.1 for each of the 4 functional platelet responses (reading from the top; PA, PC, FA, and FC). Arrow indicates the SNP with the highest association in the PA response. LD structure is based on HapMap data (as described for Figure 4), and chromosomal position in Mb based on NCBI build 36 coordinates, showing Ensembl (version 49) gene structure. (B) Calcium flux (shown as area under the curve) in response to 3 concentrations of ADP (10−7, 10−6, 10−5 M) and 5 μg/mL CRP-XL. Data are shown as the mean ± for subjects homozygous for the major allele of rs17786144 (CC) (■; n = 10), heterozygous (CT) (, n = 7), or homozygous for the minor allele (TT) (□, n = 4). (C) Calcium flux (shown as time to peak calcium) in response to 3 concentrations of ADP (10−7, 10−6, 10−5 M) and 5 μg/mL CRP-XL. Data are shown as the mean ± SD for subjects homozygous for the major allele or rs17786144 (CC) ■; n = 10), heterozygous (CT) (; n = 7), or homozygous for the minor allele (TT) (□; n = 4). (D) Example of flow cytometric histogram of expression of ITPR1 protein in platelets from subjects homozygous for the major allele of rs1786144 (solid line), heterozygous (dotted line), or homozygous for the minor allele (dashed line), illustrating the lack of effect of genotype on the level of expression. (E) Expression of ITPR1 protein, measured by flow cytometry, in platelets from 10 subjects homozygous for the major allele of rs1786144 (CC), 7 heterozygotes (CT), and 4 subjects homozygous for the minor allele (TT). (ns for all).
Discussion
The action of platelets is key to both normal hemostasis and thrombotic disease, with evidence to suggest that increased platelet responsiveness may be linked to the risk of a thrombotic event.39-42 Platelet response is a variable continuous trait that appears to be both constant within an individual2-4,8-10 and inherited.11-13 Previous studies to identify QTLs for platelet function have focused on a limited number of genes encoding membrane receptors. The design of the majority of these studies has suffered from classic pitfalls such as small sample sizes, limiting the power to detect small effects, genotyping for a single or limited number of SNPs, often selected on a priori knowledge (eg, rs5918, HPA-1), reducing the accuracy of ascertainment of underlying sequence variation, and lack of evidence of robust reproducibility of the measurement of the quantitative trait.
We reasoned that most of the variation in platelet function in the population is best explained by common sequence variation in a large number of genes each exerting a small effect.31 We therefore designed a study that was powered to detect such effects and, as a consequence, have identified 68 SNPs in 15 genes that show association with the platelet response to either, CRP-XL, ADP, or both, at a P value less than .005 (Figure 2 and Table 1). Furthermore, we have shown, through additive models, that the combined effect of multiple SNPs accounts for 38% to 46% of the variation in response to CRP-XL and 13% to 16% of the variation in response to ADP (Figure 3). Evidence from the Framingham cohort suggests that around 62% of the variation in response to collagen and 44% of the variation in response to ADP is inherited.11 On this basis, the variation associated with the genes explored in the present study, which accounted for 38, 46, 16, and 13 of the FC, PC, FA, and PA responses, respectively, would account for 62%, 74%, 36%, and 30%, respectively, of the total hereditable variation in these 4 phenotypes. While the proportion of variance explained in the general population by these SNPs may be an overestimate due to selection bias, it is undoubted that this systematic approach to intensively genotype a large number of candidate genes has identified a significant proportion of the genetic variation that underlies functional variation in the 4 signaling pathways that are central to platelet function.
As previously reported, variation in GP6 has a large impact on platelet response to CRP-XL,25 accounting for up to 40% of the variation in GPVI signaling seen in this cohort.8 For the remaining 96 genes, SNPs showing an association with phenotype had a relatively modest effect, accounting for small incremental increases of between 0.5 and 3% (Figure 3). In total, 18 independent association signals in 15 loci were observed. The genes identified here encoded for a range of functional proteins including cell-surface receptors (eg, CD36, GP6, ITGA2, PEAR1, P2RY12), adaptor proteins for agonist receptors (eg, FCERG1 and GNAZ), several kinases (eg, JAK2, MA2K2, MAP2K4, MAPK14), and other intracellular signaling molecules (eg, ITPR1 and VAV3). The likely interactions of their gene products have been illustrated in Figure 7, which indicates the cellular location, the effect size of the sequence variation in the different signaling pathways, and the likely interactions of the gene products within the platelets. Most had not been studied previously by functional genomics, and all association signals, except for that in the GP6 gene, were novel, 5 of which were discovered as a result of the sequencing effort (Table 1).

Schematic overview of the functional relationship between candidate gene products in platelets. Nodes that are colored represent genes in which sequence variation correlates with platelet functional. Genes that are colored gray have been tested, but no significant association was seen. Genes depicted as unfilled nodes were not tested but are included for pathway clarification. The diameter of a colored node is inversely proportional to the log base 10 of the lowest P value associated with any one of the platelet functional quantitative traits for that gene—the larger the node, the greater the effect (PC, P-selectin expression upon CRP-XL stimulation; FC, fibrinogen binding upon CRP-XL stimulation; PA, P-selectin expression upon ADP stimulation; FA, fibrinogen binding upon ADP stimulation). Sequence variation in a gene may be associated with more than one functional index (see Figure 4, Table 1, and supplemental Table 1 for a more detailed breakdown of individual effects). An edge between nodes denotes a functional relationship between gene products, and all are based on published data that have been interpreted by an expert in platelet signaling (J.-W.A.).
Schematic overview of the functional relationship between candidate gene products in platelets. Nodes that are colored represent genes in which sequence variation correlates with platelet functional. Genes that are colored gray have been tested, but no significant association was seen. Genes depicted as unfilled nodes were not tested but are included for pathway clarification. The diameter of a colored node is inversely proportional to the log base 10 of the lowest P value associated with any one of the platelet functional quantitative traits for that gene—the larger the node, the greater the effect (PC, P-selectin expression upon CRP-XL stimulation; FC, fibrinogen binding upon CRP-XL stimulation; PA, P-selectin expression upon ADP stimulation; FA, fibrinogen binding upon ADP stimulation). Sequence variation in a gene may be associated with more than one functional index (see Figure 4, Table 1, and supplemental Table 1 for a more detailed breakdown of individual effects). An edge between nodes denotes a functional relationship between gene products, and all are based on published data that have been interpreted by an expert in platelet signaling (J.-W.A.).
In the present study, ITGB3 showed no association with any of the markers of platelet response, despite being heavily tagged, with 49 SNPs, including all functional and HCNCR SNPs and the widely studied causal nsSNP rs5918, which underlies the HPA-1 system. There is conflicting evidence in the literature of the association of rs5918 with the platelet response.14-17,29,30 This could be due to the sample size of earlier studies, which, in sharp contrast with this study, have generally been too small to ascertain small effects of a SNP with a MAF of 0.15 (MAF in this study was 0.186, because of enrichment of HPA-1b1b individuals in the PFC).
The emergence of PEAR1 as a regulator of both CRP-XL and ADP signaling is one of the major findings of this work. Two distinct and independent associations were observed, 1 for rs41299597 (MAF = 0.017) associated with FC and PC, and another more common SNP (rs11264579, MAF = 0.190) for ADP. The PEAR1 gene has only recently been described in platelets and endothelial cells.43 Herrera-Galeano et al recently reported an association between rs2768759, a SNP 10 kb upstream of PEAR1 that maps to the NTRK1 gene, and platelet aggregation in response to collagen and epinephrine in a white American cohort and to ADP and epinephrine in a black American cohort.44 Neither this SNP, nor SNPs that are in LD, were included in the genotyping. However, subsequent genotyping of rs2768759 by TaqMan showed no association with any of the functional phenotypes measured here (data not shown). These divergent results may be due to differences in the populations studied, or in the methods used to ascertain the functional phenotype. Interestingly however, it was postulated by Herrera-Galeano et al44 that increased expression of PEAR1 might be an important cause of hyperactivity. Here, we have demonstrated that genetic variation within PEAR1, particularly rs41299597, which codes for the S802C amino acid substitution in a potential phosphorylation site in the cytoplasmic domain,43 is associated with an increased expression of PEAR1 in activated platelets as well as increased responsiveness through the GPVI receptor. That 2 of the 4 SNPs in PEAR1 were identified as a result of exon sequencing highlights the importance of the identification of rare SNPs in genotype-phenotype studies. The combination of the low MAF of SNP rs41299597 and the sample size of the PFC prevented us from unequivocally concluding that the S807C is the causative event that is the basis of the changes in the 2 functional traits, but we consider it to be highly likely.
The unique design of this genotype-phenotype association study, performed simultaneously on 4 tightly defined pathway-specific platelet phenotypes, allowed us to go beyond identifying 17 novel QTLs that regulate platelet function, to using the consequences of genetic variation (ie, changes in protein abundance or function) to study the primary physiologic role of proteins within platelets. For example, rs17229705 in VAV3, was found to be associated with increased P-selectin expression only in response to ADP. Platelets from subjects carrying the minor allele also had significantly higher VAV3 protein levels (Figure 5C). These observations suggest that the platelet response to GPVI stimulation is unaffected by variation in VAV3 protein levels, a finding that corroborates the previously demonstrated redundancy in this pathway.45 The association of VAV3 variation solely with PA is surprising, but might be explained by the role of VAV3 in regulating outside-in signaling through αIIbβ3 and the polymerization and reorganization of actin filaments.46,47 The effect of VAV3 genotype on these processes was not specifically tested in this study, however, a degree of outside-in signaling, due to the binding of fibrinogen to the αIIbβ3, is likely to contribute to the response measured in the whole blood flow cytometric assay used here. Furthermore the reinforcement of degranulation through outside-in signaling is likely to be greater in the ADP-treated samples, as activation via the GPCRs P2Y1 and P2Y12 provides a much weaker stimulus for degranulation than platelet activation via GPVI by CRP-XL.48
A similarly intriguing finding from this study concerns ITPR1, which codes for the IP3 receptor, that plays a key role in the stimulus-mediated release of calcium from the endoplasmic reticulum.49 In platelets, this ion channel has been shown to be regulated by IP3 released by both PLCγ2 and PLCβ, activated, respectively by the GPVI receptor complex or by GPCRs. The observation that the effect of rs170414 was restricted solely to the ADP signaling pathway is of interest. Whether it suggests that this polymorphism is more apparent at lower levels of IP3 that may be released as a consequence of the relatively weak stimulus caused by ADP, or it reflects differential effects of PLCγ2 and PLCβ on the generation of IP3 is unclear. It would be interesting to see whether this SNP also affects the response to a stronger agonist that operated through a PLCβ-mediated, GPCR pathway, such as thrombin or thromboxane.
In this study, we have identified a large proportion of the QTLs regulating 4 functional platelet traits. We have used a systematic approach and novel design in which genetic variation is not just identified as an end point, but is also used to inform the physiologic relevant action of proteins within intracellular signaling pathways. As such, this represents the first step in an effort to systematically unravel the complex relationship between sequence variation and platelet functional phenotype using high-throughput genotyping of a large number of genes. Building on the successful demonstration of this strategy, future work using genome-wide genotyping arrays complemented by higher resolution tagging of strategies of candidate regions will identify novel loci, and the measurement of a wider range of tightly defined phenotypes will radically enhance our understanding of both the effect of genetic variation on the proteins they encode and how these proteins interact within intracellular signaling pathways to regulate cellular function. Finally, QTLs identified in this study can be used in genotyping studies of cases of MI or stroke to determine whether they confer risk of disease.
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the following: the staff of the Donor Clinic of National Health Service Blood and Transplant (NHSBT) in Cambridge for invaluable assistance in obtaining donor samples; Dr Nicolas Raynal (Department of Biochemistry, University of Cambridge) for the CRP-XL; Rosey Mushens and colleagues of Bristol Institute for Transfusion Sciences (BITS), Bristol, United Kingdom for the PE-CD62P antibody; Dr Mark L. Kahn (Department of Medicine, University of Pennsylvania) for the HY101 anti-GPVI; and Steve Darwood, Martin Dominguez, and colleagues at Beckman-Coulter Ltd for help in setting up automated data transfer of the flow cytometric data. We also gratefully acknowledge the generosity of the healthy blood donors who have made their DNA samples available to this project as a “free gift.”
The Bloodomics project (www.bloodomics.org) is funded by the 6th Framework Program of the European Union (LSHM-CT-2004-503485). The project made use of volunteers from the Cambridge BioResource, a Cambridge Biomedical Research Centre resource funded by the National Institute for Health Research (NIHR).
Authorship
Contribution: C.I.J. performed the platelet function research, participated in the design of the research and analysis of the data, and wrote the paper; S.B. analyzed the data and helped write the paper; S.F.G. performed the platelet function research and helped write the paper; J.S. performed the platelet function research; B.d.B. contributed bioinformatical analysis of the data and generated Figure 7; W.G.J.A. contributed vital analytical tools and bioinformatics support; D.B. participated in the design of the research; P.B. performed the transcriptomic research; A.C. performed the resequencing; P.D. participated in the design of the research and provided vital resources for genetic studies; M.E. performed the resequencing; R.W.F. participated in the design of the research, provided vital reagents, and contributed vital expertise in platelet signaling; M.F.H. participated in the design of the research; K.K. contributed vital analytic and bioinformatical support; A.R. performed the platelet function research; C.M.R. contributed vital bioinformatical tools and support; J.R. participated in the design of the research and oversaw exon resequencing; N.J.S. participated in the design of the research; M.S. contributed vital new reagents; A.W. contributed vital new reagents; N.A.W. participated in the design of the research and oversaw transcriptome analysis; J.-W.A. participated in the design of the research and contributed vital expertise in platelet signaling; F.D. analyzed the data and helped write the paper; A.H.G. designed the research and wrote the paper; W.H.O. designed the research and wrote the paper; and the Bloodomics Consortium provided vital infrastructure and expertise to the research.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
For a complete list of Bloodomics Consortium participants, see the supplemental Appendix.
Correspondence: Alison H. Goodall, Department of Cardiovascular Sciences, University of Leicester, Clinical Sciences Wing, Glenfield Hospital, Leicester, LE3 9QP, United Kingdom; e-mail: ahg5@le.ac.uk.

 , n = 7), or homozygous for the minor allele (TT) (□, n = 4). (C) Calcium flux (shown as time to peak calcium) in response to 3 concentrations of ADP (10−7, 10−6, 10−5 M) and 5 μg/mL CRP-XL. Data are shown as the mean ± SD for subjects homozygous for the major allele or rs17786144 (CC) ■; n = 10), heterozygous (CT) (
, n = 7), or homozygous for the minor allele (TT) (□, n = 4). (C) Calcium flux (shown as time to peak calcium) in response to 3 concentrations of ADP (10−7, 10−6, 10−5 M) and 5 μg/mL CRP-XL. Data are shown as the mean ± SD for subjects homozygous for the major allele or rs17786144 (CC) ■; n = 10), heterozygous (CT) (