

Abstract
Abstract 4783
Polycythemia/Erythrocytosis may be congenital and/or familial, or acquired. The first recognized familial polycythemias are associated with mutations in the globins gene resulting in increased affinity of Hb to oxygen, which could also be caused by congenital deficiency of 2, 3 DPG. Subsequently gain-of-function of erythropoietin receptor gene mutations have been shown to cause familial dominantly inherited polycythemia. More recently, mutation in the genes involved in hypoxia sensing pathway have been described as causes of congenital polycythemia; these include recessively inherited VHL (Chuvash polycythemia), and dominantly inherited HIF2a and PHD2 mutations. Yet unexplained familial polycythemias still exist. Here we investigated a family with dominant polycythemia present in five generations wherein all known mutations in polycythemia-causing genes were excluded through linkage analyses and direct sequencing. Familial clustering of the disease provided us an opportunity to identify a novel genetic cause modulating erythropoiesis. We utilized a shared genomic segment (SGS) and linkage analysis approach, to provide evidence of chromosomal candidate regions for further efforts to localize the causative gene.
A five generation family pedigree consisting of 26 phenotyped members (10 affected/16 unaffected) with 16 subjects (5 affected and 11 unaffected) genotyped on an Affymetrix v5.0 SNP platform. A conventional two-point linkage analysis did not identify a potential causative genomic region. Multipoint linkage analysis was subsequently performed because this allows for optimal use of the genetic SNP data to infer inheritance patterns and leads to lower false positive rates than two-point linkage methods. In order to identify potential causative regions for harboring the polycythemia-causing defect, we also performed shared genomic segment (SGS) analysis that recognizes a region identical by descent must also be shared by identical by state. We analyzed 409,426 reference dbSNPs with a 94% call rate for genotyped subjects to determine runs of loci at which affected individuals share a common allele. Parametric multipoint linkage analysis used a pruned set of 8,394 non-monomorphic markers with minimum spacing of 100 kb, minimum heterozygosity of 0.3 and maximum pairwise r2 of 0.16 based on HapMap CEU data. We used a Markov chain Monte Carlo linkage method (MCLINK) which allows for fully informative multilocus linkage analysis on extended pedigrees.
SGS identified five runs of shared loci that achieved chromosome-wide significance among all five affected subjects (chromosome, run length): 18, 1980 SNPs; 14, 1292 SNPs and 1095 SNPs; 16, 1060 SNPs and 20, 500 SNPs. The SGS regions on chr. 14 (1292 and 1095 run lengths out of 13,149 total SNPs) begins at (NCBI build 36.1) 55,956,582,765bp (rs17254216) through 73,408,302bp (rs1778222) and contain regions of 80 and 79 consecutive SNPs at which all cases share homozygously from a homozygous shared genomic segment analysis (HGS). Multipoint linkage identified a peak LOD score of 3.41 on chr. 16 at 78,033,240bp (rs4888031) achieving genomewide significance which is contained within an observed SGS region of interest (77,507,467bp to 81,049,285bp).
Summary of SGS, HGS, and linkage evidence for the Pedigree with PFCP.
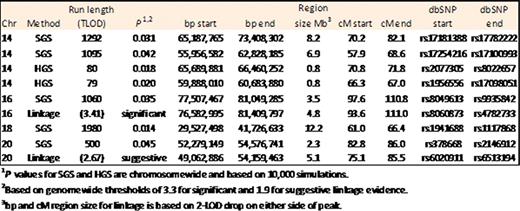
We submit that genetic epidemiological analyses (SGS and Linkage) augment likelihood of identifying the disease-causing genomic regions that are good candidates for the underlying familial susceptibility variant(s). We observed an 8Mb region of interest on chromosome 14 which may contain a segment of loss-of-heterozygosity, and a 3.9Mb region on chromosome 16 both are now being analyzed by sequencing studies.
No relevant conflicts of interest to declare.

