

Abstract
Abstract 5172
Sickle cell disease (SCD) and β thalassemia due to defects of the HBB (β globin gene) are among the most common inherited genetic disorders. At birth, there is a switch of γ globin transcription to β and d, with replacement of HbF by HbA and HbA2 virtually completed by six months of age. At that time, serious inherited disorders of the β gene, such as sickle cell disease and Cooley's anemia (homozygosity for β0 thalassemia mutations), become clinically apparent. Cooley's anemia is a life-threatening disorder wherein, in most patients, chronic transfusions or bone marrow transplantations are needed to sustain life. Rare patients with homozygosity or compound heterozygosity for β0 have no or only mild anemia. These patients maintain a high level of γ globin synthesis, apparently from a disrupted γ-to-β switch, thus attenuating their disease state. Recent work has demonstrated that BCL11A plays an important role in the suppression of γ-globin expression, as do polymorphisms of the gene that remain to be fully elucidated at a functional level. We recruited two unrelated subjects with homozygous β0 thalassemia mutations with no or only mild anemia (Patient #1, IVS2+1 G>A; Hb 14.2 Gm%; 97.2% HbF, 2.8% HbA2, Patient #2, IVS 2 G-T; Hb 11.2 Gm%; 92/5% HbF, 6.8% HbA2, 0.7% HbA). We sequenced transcripts and genomic loci of BCL11A from these patients. No mutations or splicing variants on transcripts were found. However, when the ≂f102 kb of genomic material from patient #1 was sequenced, 5 single nucleotide changes at intron II were found (2 known and 3 previously unpublished), while no genomic changes were found in patient #2. We then performed in vitro erythroid expansion from peripheral blood utilizing high erythropoietin concentrations and analyzed the cell proliferation and expression of globin and BCL11A genes. Interestingly, detectable amounts of β-globin transcripts were present in both patients during expansion, although protein levels were not detectable by the conventional HPLC method, probably due to limited sensitivity of this assay. Patient #1 showed mild in vitro induction of β-globin expression, which is lower than the control group, but no apparent cell proliferation. Patient #2 showed no induction of β-globin expression and hyperproliferation at a later stage of expansion (See Figure); however, the levels of BCL11A and γ-globin transcripts were indistinguishable from controls.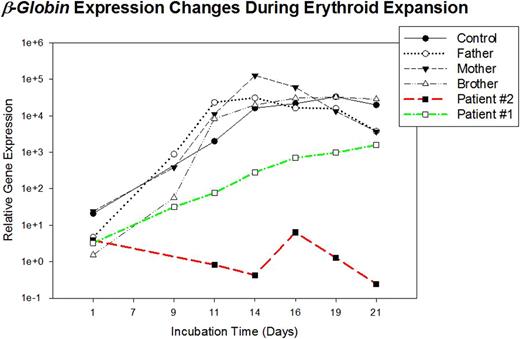

Although we were unable to detect any abnormality of the BCL11A transcript as a cause of high fetal hemoglobin expression in these patients, we cannot rule out the possibility that the intronic mutations in patient #1 may interfere with BCL11A gene translation, perhaps by interference with non-coding RNA. The potential molecular mechanism of γ-to-β switch is being explored by gene expression profiling and microRNA analyses.
Disclosures:
No relevant conflicts of interest to declare.

