

Abstract
Protein S (PS) enhances the inhibition of factor Xa (FXa) by tissue factor pathway inhibitor-α (TFPI-α) in the presence of Ca2+ and phospholipids. Altered forms of recombinant TFPI-α were used to determine the structures within TFPI-α that may be involved in this PS-dependent effect. Wild-type TFPI-α (TFPIWT), TFPI-α lacking the K3 domain (TFPI-ΔK3), and TFPI-α containing a single amino acid change at the putative P1 residue of K3 (R199L, TFPIK3P1) produced equivalent FXa inhibition in the absence of PS, whereas the response in FXa inhibition produced by PS was reduced with TFPIK3P1 (EC50 61.8 ± 13.4nM vs 8.0 ± 0.4nM for TFPIWT) and not detectable with TFPI-ΔK3. Ligand blotting and surface plasmon resonance experiments demonstrated that FXa bound TFPIWT and TFPI-ΔK3 but not the isolated K3 domain, whereas PS bound TFPIWT and the K3 domain but not TFPI-ΔK3. Addition of TFPIWT, TFPIK3P1, or TFPI-ΔK3 produced comparable prolongation of FXa-induced coagulation in PS-deficient plasma, but the anticoagulant effect of TFPIWT was substantially greater than that of TFPIK3P1 > TFPI-ΔK3 in normal plasma and PS-deficient plasma reconstituted with PS. We conclude that the PS-mediated enhancement of FXa inhibition by TFPI-α involves an interaction between PS and TFPI-α, which requires the K3 domain of TFPI-α.
Introduction
Factor X (FX) is a vitamin K–dependent blood coagulation zymogen that plays a central role in hemostasis.1,2 Activated FX (FXa) assembles with its cofactor FVa in the prothrombinase complex FXa/FVa/Ca2+/phospholipids (PLs) to catalyze the conversion of prothrombin to thrombin.3 The physiologic activation of FX is catalyzed by FVIIa/tissue factor (TF)/Ca2+/PL (extrinsic Xase complex) or FIXa/FVIIIa/Ca2+/PLs (intrinsic Xase complex).3,4 The activation of FX catalyzed by the extrinsic Xase complex proceeds through an initial formation of a FVIIa/TF/FX ternary complex, followed by cleavage of FX by FVIIa (FVIIa/TF/FXa) and subsequent release of FXa. This mode of FXa generation is tightly regulated by tissue factor pathway inhibitor-α (TFPI-α), a trivalent Kunitz-type protease inhibitor synthesized predominantly by endothelial cells.5,6
TFPI-α is a glycoprotein of 276 amino acid residues organized into an acidic N-terminal sequence, 3 tandem Kunitz-type inhibitory domains denoted Kunitz-1 (K1), Kunitz-2 (K2), and Kunitz-3 (K3), respectively, and a basic C-terminal tail.7 TFPI-α inhibits FVIIa/TF and FXa simultaneously by binding to the FVIIa/TF/FXa complex to form a FVIIa/TF/FXa/TFPI-α tetramolecular complex.8 In this complex, the K1 domain of TFPI-α binds to the active site of FVIIa, whereas the K2 domain binds to the active site of FXa.9 The role of the K3 domain of TFPI-α, which lacks proteinase inhibitory activity,10 has been obscure, but recent studies have shown it to be involved in the association of TFPI-α with cell surfaces.11
TFPI-α is also a potent inhibitor of FXa, independent of the FVIIa/TF complex. Recently, it was observed that, in the presence of Ca2+ and PLs, direct TFPI-α inhibition of FXa is significantly enhanced by protein S (PS),12 a vitamin K–dependent protein better known for its role as a cofactor for activated protein C in the inactivation of FVa and FVIIIa. The mechanism by which PS promotes TFPI-α inhibition of FXa is not completely understood. Previous studies showed that the inhibition of FXa by a truncated form of TFPI (TFPI1-161) that contained only the N-terminal and K1 and K2 domains of TFPI-α was not affected by PS.12,13 This suggested that a structure(s) in TFPI-α down-stream of the K2 domain is required for the enhancement in FXa inhibition produced by PS.
Methods
Proteins and reagents
Bovine serum albumin (BSA) was purchased from Sigma-Aldrich. FVa and prothrombin were from Haematologic Technologies. Human α-thrombin, PS, FXa, and PS-deficient plasma were from Enzyme Research Laboratory. Pooled normal plasma was from George King Biomedical. H-D-Phe-Pip-Arg-p-nitroanilide (S-2238) and benzoyl-Ile-Glu-Gly-Arg-p-nitroanilide (S-2222) were from Diapharma. PL in the form of rabbit brain cephalin was purchased from Centerchem. Anti-TFPI K2 domain monoclonal antibody (Mab2B12) and anti-TFPI K1 domain monoclonal antibody (Mab2H8) have been previously described.14 A polyclonal antibody developed in rabbit against the peptide (KIAYEEIFVKNM, TFPI265-276), which corresponds to the C-terminus of TFPI-α, has also been described.15
Expression and purification of TFPI proteins
A plasmid, pUCGB9R1, containing the TFPI-α cDNA modified for insertion into a mammalian expression vector16 and standard techniques were used16,17 to generate the following altered forms of rTFPI: (1) rTFPI1-252 (TFPIC1), lacking one-half of the C-terminus; (2) rTFPI1-242 (TFPIC2), lacking the entire C-terminus; (3) rTFPI1-181 (TFPIC3), lacking both the K3 domain and C-terminus; (4) rTFPIdes181-243 (TFPI-ΔK3), K3 domain deleted; and (5) rTFPIR199L (TFPIK3P1), a point mutation at the putative P1 site of the K3 domain (Figure 1). Stable clones of C127 mouse mammary tumor cells expressing wild-type and the mutant forms of rTFPI were produced as previously described.11
The recombinant WT and altered TFPI proteins were purified from serum-free conditioned medium by immunoaffinity chromatography as described,15 with minor modifications. Briefly, media were cleared of cell debris by centrifuging for 1 hour at 4000g, then filtered through a 0.22-μm GP Express PLUS membrane (Millipore), and applied to a Mab2H8 monoclonal anti–TFPI-agarose column by gravity flow at 4°C. The column was washed with Tris-buffered saline (TBS; 50mM Tris, 150mM NaCl, pH 7.4) and eluted with 0.1M glycine, pH 2.2. The pH of the eluted fractions was adjusted to 7.5 using 1M Tris, pH 8. Fractions were assayed for TFPI activity by the inhibition of FXa amidolytic activity using the chromogenic substrate S-2222. Fractions with the highest activities were pooled, dialyzed against TBS, and concentrated using the Centricon Centrifugal Filter Devices (YM 30; Millipore). Further purification of TFPIWT, TFPIK3P1, and TFPI-ΔK3 was carried out by heparin-agarose chromatography as described.15 The purified TFPI proteins were stored at −80°C until use.
The 2-domain TFPI (TFPI161), which consists of amino acids 1 to 161 of the full-length molecule and contains the N-terminal sequence, the K1 and K2 domains, but lacks the K3 domain and C-terminal peptide, was expressed in Escherichia coli, purified, and renatured as described elsewhere.18 To express the K3 domain, the DNA fragment corresponding to residues 182 to 241 of TFPI-α was generated by polymerase chain reaction from TFPI cDNA with primers 5′-ATCTCGAGGAATTTCACGGTCCCTCA and 5′-GCAAGCTTCTATTLTTTTACATGCCCTC, which include XhoI and HindIII sites. The amplified fragment was cloned into vector pRSET A (Invitrogen) XhoI and HindIII sites. The plasmid was then transformed into E coli host BL21(DE3). A 1-L culture was grown to an optical density (A600) of approximately 0.6 and then induced with 1mM isopropyl β-D-1-thiogalactopyranoside for 4 hours. The bacteria were pelleted, and lysed by sonication in a TBS (20mM Tris-HCL, pH 8.0, with 500mM sodium chloride) containing 50mM imidazole, followed by centrifugation at 16 000g for 30 minutes. Soluble K3 protein in the supernatant was then isolated by cobalt affinity followed by anti-poly His monoclonal antibody (Sigma-Aldrich) affinity chromatography. The purified protein was more than 90% pure as judged by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The protein concentration was determined by the Bio-Rad protein assay using known concentration of TFPI161 as standard.
TFPI immunoassay
The concentrations of the purified proteins were determined by sandwich enzyme-linked immunosorbent assay using Mab2B12 and biotin-conjugated Mab2H8 as described,19 with minor modifications. Briefly, wells of high binding 96-well plates (Fisher Scientific) were incubated overnight at room temperature with 100 μL of Mab2B12 (20 μg/mL in TBS), and then washed 5 times with TBS containing 0.05% Tween 20 (TBST). All subsequent incubation steps were carried out at room temperature. TFPI samples (100 μL) were applied to the wells and incubated for 2 hours. Thereafter, wells were washed 5 times with TBST and 100 μL of biotin-conjugated Mab2H8 (4 μg/mL) was applied and incubated for 1.5 hours. The wells were washed 5 times with TBST, and 100 μL of strepavidin-horseradish peroxidase complex (1 μg/mL, Pierce Chemical) in TBST was applied. After 1 hour, the wells were washed 5 times with TBST and 100 μL of 3,3′,5,5′-tetramethylbenzidine liquid substrate system (Sigma-Aldrich) was added. After 5 minutes, 50 μL of 0.5M H2SO4 was added to stop the reaction, and the absorbance at 450 nm was measured with a Vmax microtitre plate reader (Molecular Devices). Purified recombinant TFPI expressed in E coli20 was used as the standard and produced a linear response between concentrations of 0 and 0.6nM.
Inhibition of FXa by purified TFPI proteins in the absence of PS
All FXa inhibition reactions were carried out at room temperature in microtiter plate format using a total volume of 160 μL and TBS/BSA buffer (TBS containing 0.1% BSA). Increasing concentrations of TFPI (0-16nM) were premixed with S-2222 (500μM), CaCl2 (5mM), and PL (35μM). FXa (200pM) was then added to start the reaction. S-2222 hydrolysis was followed by continuous measurement of the absorbance at 405 nm (p-nitroaniline release) in a Vmax microtiter plate reader (Molecular Devices). Data were analyzed based on the slow, tight-binding mechanism, using the following equation21-23 :
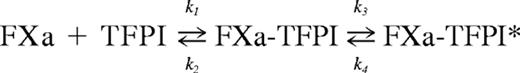
where FXa-TFPI is the initial encounter complex that isomerizes to a tightened FXa-TFPI* form. The constants k1 and k2 are the forward and reverse rates for the initial binding, and k3 and k4 are the forward and reverse rates for the isomerization step, respectively. The steady-state velocity (vs) of S-2222 hydrolysis was calculated by fitting the data to the following equation, using the Grafit program (Version 3.01) from Erithacus Software (Sigma-Aldrich):
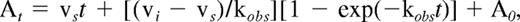
where At and A0 represent absorbance at 405 nm at time t and time 0 (background value), respectively. The observed rate constant, kobs, is the rate of conversion of the initial velocity (vi) of S-2222 hydrolysis to vs.
Effect of protein S on TFPI inhibition of FXa
For these reactions, a fixed amount of TFPI (4nM) and various concentrations of PS (0-320nM) were mixed before the addition of S-2222 (500μM), CaCl2 (5mM), and PL (35μM). FXa (400pM) was then added to start the reaction. FXa activity (S-2222 hydrolysis) was followed and the data analyzed as described for the inhibition of FXa by purified TFPI proteins in the absence of PS in the previous section. To determine the EC50 values (concentration of PS yielding 50% of the maximal potentiation of TFPI inhibition of FXa), the final velocities of S-2222 hydrolysis (vs) at each PS concentration were fitted to the IC50 4-parameter logistic equation of Halfman,24 given below.
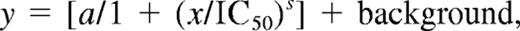
where y is the rate of S-2222 hydrolysis in the presence of a given concentration of PS represented by x, a is the maximum rate of S-2222 hydrolysis in the absence of PS, and s is the slope factor.
Binding of TFPI to PS in 96-well plate
Wells of high binding 96-well plate (Fisher Scientific) were incubated with either 100 μL of PS (100 μg/mL) in 50mM carbonate buffer (1:2 of Na2CO3/NaHCO3), pH 9.6, or plain buffer overnight at 4°C. All subsequent steps were carried out at room temperature. The plate was blocked with 3% BSA in TBST for 1 hour and then washed 5 times with TBST. Next, 100 μL of TFPI samples (2.5nM) in duplicates was applied to the wells and incubated for 2 hours. The wells were washed 5 times with TBST and the bound TFPI detected with the biotin-conjugated Mab2H8 as described in “TFPI immunoassay.” Specific binding was obtained by subtracting the background signals from wells coated with buffer only from the corresponding wells coated with PS.
Binding of PS to TFPI using SPR
Studies were performed on a Biacore T100 instrument (GE Healthcare). Each TFPI (100nM in 10mM sodium acetate, pH 4.5) was immobilized on a carboxymethyldextran-modified gold surface (CM5) chip at a flow rate of 5 μL/minute via a standard 1-ethyl-3-(3-dimethyl-aminopropyl) carbodiimide/N-Hydroxy Succinamide (EDC/NHS) –mediated amine coupling protocol.25 This was followed by a 3-minute pulse of 1M ethanolamine to deactivate the excess ester groups. The immobilization level for each TFPI was approximately 2000 RU. PS binding experiments were performed at 25°C using TBS containing 5mM CaCl2 and 0.005% Tween 20. PS at different concentrations (0-4000nM) was injected for 5 minutes at a flow rate of 30 μL/minute, and dissociation was monitored for 5 minutes. Surface regeneration between experiments was achieved with a 5-minute pulse of 1M NaCl in running buffer. Response data were processed using 1 flow cell coupled only with ethanolamine as a reference surface to correct for bulk refractive index changes and nonspecific binding.
Ligand and Western blotting
For ligand blotting with PS, TFPI proteins were electrophoresed on 12% SDS-PAGE using the Laemmli buffer system26 and blotted onto a 0.45-μm protran pure nitrocellulose transfer and immobilization membrane (PerkinElmer Life and Analytical Sciences). The membrane was blocked with 5% fat-free milk in TBST at room temperature for 1 hour and then incubated with PS (4 μg/mL) in 1% milk in TBST overnight at 4°C. After 3 washes with TBST, the membrane was incubated with polyclonal rabbit anti–human PS antibody (Dako North America) at room temperature for 1 hour. A second antibody, horseradish peroxidase-goat anti–rabbit IgG (Sigma-Aldrich), and the Lumi-LightPlus Western blotting substrate kit (Roche Diagnostics) were used to detect the primary polyclonal antibody. For ligand blotting with FXa, the same membrane used for PS blotting described in the preceding procedure was stripped by incubating with 100mM glycine, pH 2.2, at room temperature for 20 minutes. After 3 washes with TBST, the membrane was incubated for 1 hour at room temperature with FXa (1 or 0.1 μg/mL). After 3 washes with TBST, the membrane was incubated with polyclonal goat anti–human FX antibody (Haematologic Technologies) at room temperature for 1 hour. A second antibody, horseradish peroxidase-rabbit anti–goat IgG (Sigma-Aldrich) and the Lumi-LightPlus Western blotting substrate kit were used to detect the primary antibody. For Western blotting, TFPI proteins were electrophoresed and blotted as described for ligand blots earlier in this paragraph. The blot was then probed with a rabbit polyclonal antibody against the C-terminal peptide of TFPI-α and subsequently detected with the horseradish peroxidase-goat anti–rabbit IgG as described for the detection of PS in ligand blotting.
Effect of PS and TFPI on prothrombinase activity
Reactions were performed at room temperature in TBS/BSA buffer. The concentrations of reactants listed in the procedure that follows represent final concentrations. The prothrombinase complex (10pM FXa, 10nM FVa, 5mM CaCl2, 35μM cephalin) was constructed and added to mixtures containing prothrombin (1μM) with or without TFPI-α (1nM) and with or without PS (200nM). Samples were withdrawn at various times and diluted into TBS containing ethylenediaminetetraacetic acid (20mM) to stop the reaction. S-2238 (100μM) hydrolysis was followed by continuous measurement of the absorbance at 405 nm (p-nitroaniline release) for 5 minutes. The thrombin concentration was determined using a standard curve generated with known amounts of human α-thrombin.
FXa-induced clotting assay
FXa-induced coagulation was performed as previously described27 with minor modifications. Briefly, TFPI diluted in TBS/BSA (50 μL) was mixed with 50 μL of pooled normal plasma, PS-deficient plasma, or PS-deficient plasma spiked with 200nM PS in a fibrometer cup. A second mixture (150 μL) composing FXa, CaCl2, and cephalin diluted in TBS/BSA was also constructed. Both mixtures were allowed to preincubate separately at 37°C for 3 minutes, after which the FXa/CaCl2/cephalin mix was transferred to the TFPI/plasma mix in the fibrometer (BBL), and the clot times recorded. Final concentrations for each reagent were TFPI (0-20nM), FXa (2nM), cephalin (25μM), and CaCl2 (5mM). Assays were performed in duplicate.
Results
To identify the structure(s) in TFPI-α needed for the PS-dependent enhancement of FXa inhibition, altered forms of TFPI-α (Figure 1) were produced and their inhibition of FXa examined in the presence and absence of PS. The P1 residue in inhibitory Kunitz domains plays a critical role in proteinase binding and inhibition. Although the K3 domain of TFPI-α does not possess proteinase inhibitory activity,10 a form of TFPI-α in which only the putative P1 residue of K3 was altered (TFPIK3P1) was also constructed to assess whether this residue could be involved in a PS–TFPI-α interaction.

Schematic of altered TFPI forms. The dark line represents the TFPI sequence; and the gray ovals, the Kunitz domains. Numbers in italics indicate the residue at the sites of truncations. Note that TFPIK3P1 is a point mutation.
Schematic of altered TFPI forms. The dark line represents the TFPI sequence; and the gray ovals, the Kunitz domains. Numbers in italics indicate the residue at the sites of truncations. Note that TFPIK3P1 is a point mutation.
Inhibition of FXa by TFPI in the absence of PS
Progress curves of FXa inhibition in the presence of Ca2+ and PL by TFPIWT and the altered forms of TFPI are presented in Figure 2. These data show that TFPI-ΔK3 and TFPIK3P1, which possess an intact C-terminus, have anti-FXa activities comparable with that of TFPIWT. For example, the steady-state rates of S-2222 hydrolysis by FXa in the presence of 4nM TFPI (A405/minute, from Figure 2) are TFPIWT (0.63 mOD/minute) TFPI-ΔK3 (0.61 mOD/minute), and TFPIK3P1 (0.93 mOD/minute). In contrast, the TFPI forms with truncated C-termini (TFPIC1, TFPIC2, or TFPIC3) have reduced FXa inhibitory activities. For these TFPI forms, the steady-state rates of S-2222 hydrolysis by FXa in the presence of 4nM TFPI are TFPIC1 (2.06 mOD/minute), TFPIC2 (1.78 mOD/minute), and TFPIC3 (1.82 mOD/minute), all of which are approximately 3-fold higher than the rate in the presence of TFPIWT. These findings are consistent with our previous report that the C-terminus of TFPI is required for the optimal inhibition of FXa.15

Inhibition of FXa by the TFPI forms in the absence of PS. Reactions were performed in TBS/BSA. Progress curves for the hydrolysis of S-2222 by FXa as measured by ΔA405 nm are presented at different concentrations of each TFPI. Reaction mixtures in 160 μL contained S-2222 (500μM), CaCl2 (5mM), cephalin (35μM), FXa (200pM), and TFPI (0-16nM). For all panels, the concentrations of TFPI from top to bottom are 0, 0.5, 1, 2, 4, 8, and 16nM, except for TFPIC2 where the highest concentration is 12nM and not 16nM. The data were fitted by nonlinear regression using the program Grafit (Version 3.01) from Erithacus software. Each experiment was repeated at least once. One set of experiments is shown.
Inhibition of FXa by the TFPI forms in the absence of PS. Reactions were performed in TBS/BSA. Progress curves for the hydrolysis of S-2222 by FXa as measured by ΔA405 nm are presented at different concentrations of each TFPI. Reaction mixtures in 160 μL contained S-2222 (500μM), CaCl2 (5mM), cephalin (35μM), FXa (200pM), and TFPI (0-16nM). For all panels, the concentrations of TFPI from top to bottom are 0, 0.5, 1, 2, 4, 8, and 16nM, except for TFPIC2 where the highest concentration is 12nM and not 16nM. The data were fitted by nonlinear regression using the program Grafit (Version 3.01) from Erithacus software. Each experiment was repeated at least once. One set of experiments is shown.
Inhibition of FXa by TFPI in the presence of PS
Data for the inhibition of FXa in the presence of Ca2+ and PL by a fixed concentration of each TFPI with various concentrations of PS are shown in Figure 3. Increasing concentrations of PS progressively enhance the inhibition of FXa by TFPIWT, TFPIC1, and TFPIC2 with apparent saturation achieved at approximately 160nM PS (Figure 3A top panel). In contrast, PS had no effect on the inhibition of FXa by TFPIC3 and TFPI-ΔK3, and the response to PS of TFPIK3P1 was significantly reduced compared with that of TFPIWT (Figure 3A bottom panel). The final rates of S-2222 hydrolysis by FXa in the presence of the various TFPI forms and at different PS concentrations (from Figure 3A) are plotted in Figure 3B. The EC50 values for PS derived from Figure 3B are shown in Table 1. PS modestly inhibited the inactivation of FXa by TFPI-α with or without Ca2+ in the absence of PL (data not shown).

Inhibition of FXa by the TFPI forms in the presence of protein S. (A) Progress curves for S-2222 hydrolysis by FXa in the presence of TFPI and various concentrations of PS. Final concentrations were S-2222 (500μM), TFPI (4nM), FXa (400pM), CaCl2 (5mM), and cephalin (35μM). For each dataset, the concentrations of PS from top to bottom are 0, 5, 10, 40, 80, 160, and 320nM. The data were fitted by nonlinear regression using the program Grafit (Version 3.01) from Erithacus software as described.21-23 (B) Plot of the final rates of S-2222 hydrolysis data from Figure 3A fitted into the IC50 4-parameter logistic equation of Halfman.24 Note that, in the absence of PS (0nM PS), TFPIWT, TFPI-ΔK3, and TFPIK3P1 inhibit FXa better, hence the lower A405/minute values compared with TFPIC1, TFPIC2, and TFPIC3.
Inhibition of FXa by the TFPI forms in the presence of protein S. (A) Progress curves for S-2222 hydrolysis by FXa in the presence of TFPI and various concentrations of PS. Final concentrations were S-2222 (500μM), TFPI (4nM), FXa (400pM), CaCl2 (5mM), and cephalin (35μM). For each dataset, the concentrations of PS from top to bottom are 0, 5, 10, 40, 80, 160, and 320nM. The data were fitted by nonlinear regression using the program Grafit (Version 3.01) from Erithacus software as described.21-23 (B) Plot of the final rates of S-2222 hydrolysis data from Figure 3A fitted into the IC50 4-parameter logistic equation of Halfman.24 Note that, in the absence of PS (0nM PS), TFPIWT, TFPI-ΔK3, and TFPIK3P1 inhibit FXa better, hence the lower A405/minute values compared with TFPIC1, TFPIC2, and TFPIC3.
Binding of PS to TFPIWT
In a preliminary screen to assess the interaction between PS and TFPI, PS was found to bind TFPIWT and, to a lesser degree, TFPIC1, TFPIC2, and TFPIK3P1, but not TFPIC3 and TFPI-ΔK3, which lack the K3 domain (Figure 4A). The binding of PS to TFPIWT and TFPI-ΔK3 was further evaluated by surface plasmon resonance (SPR). With approximately 2000 RU of each TFPI coupled to adjacent flow cells of the same CM5 chip, increasing concentrations of infused PS led to a corresponding increase in the binding of PS to TFPIWT, with a response of approximately 80 RU above baseline obtained with 4000nM PS (Figure 4B). On the other hand, even at a concentration of 4000nM, binding of PS to TFPI-ΔK3 was undetectable (Figure 4C). In similar experiments, increasing concentrations of infused PS also led to a corresponding increase in the binding of PS to TFPIC1 and TFPIK3P1 (data not shown). Ca2+ alone did not enhance the binding of PS to TFPIWT as similar binding responses were observed in the presence or absence of Ca2+. In the presence of Ca2+ and PL, however, the binding of PS to TFPIWT was increased approximately 5-fold (not shown). The sensorgrams of the binding of PS to TFPIWT and other forms of TFPI-α demonstrated a complex pattern, did not reach equilibrium, and could not be adequately fit using the analytical software provided with the Biacore instrument. This prevented a reliable delineation of binding constants, but plots of PS binding at an arbitrary time point (300 seconds) suggest a dissociation constant more than 1μM for TFPIWT and greater values for TFPIK3P1 and TFPIC1. Nonetheless, the studies confirm a direct interaction between PS and TFPI-α, which is lost when the K3 domain is deleted.

Binding of PS to TFPI. (A) Microtiter plate assay. Wells of a 96-well plate were precoated with PS and then blocked with BSA. TFPI samples were added to the wells and incubated for 2 hours. Thereafter, wells were washed and the bound TFPI detected with a biotin-conjugated Mab2H8 as described under “TFPI immunoassay.” Specific binding of each form of TFPI to PS was obtained by subtracting the readings of wells coated with plain buffer (background) from the corresponding readings of wells coated with PS. The binding of TFPI to PS is expressed as a percentage of the binding obtained with TFPIWT, set at 100%. Assays were done in duplicates. (B-C) SPR. A CM5 chip flow cell was coupled with TFPI to approximately 2000 RU. Cells were then perfused with increasing concentrations of PS (0, 62.5, 125, 250, 500, 1000, 2000, and 4000nM), in TBS containing 5mM CaCl2 and 0.005% Tween 20, as described in “Binding of PS to TFPI using SPR.” The sensorgrams shown have been corrected for the signal obtained in a reference flow cell without immobilized TFPI.
Binding of PS to TFPI. (A) Microtiter plate assay. Wells of a 96-well plate were precoated with PS and then blocked with BSA. TFPI samples were added to the wells and incubated for 2 hours. Thereafter, wells were washed and the bound TFPI detected with a biotin-conjugated Mab2H8 as described under “TFPI immunoassay.” Specific binding of each form of TFPI to PS was obtained by subtracting the readings of wells coated with plain buffer (background) from the corresponding readings of wells coated with PS. The binding of TFPI to PS is expressed as a percentage of the binding obtained with TFPIWT, set at 100%. Assays were done in duplicates. (B-C) SPR. A CM5 chip flow cell was coupled with TFPI to approximately 2000 RU. Cells were then perfused with increasing concentrations of PS (0, 62.5, 125, 250, 500, 1000, 2000, and 4000nM), in TBS containing 5mM CaCl2 and 0.005% Tween 20, as described in “Binding of PS to TFPI using SPR.” The sensorgrams shown have been corrected for the signal obtained in a reference flow cell without immobilized TFPI.
In additional ligand-blotting studies, PS bound TFPIWT and the isolated TFPI K3 domain in a Ca2+ and PL-independent fashion (Figure 5A-B), whereas no detectable binding was observed to TFPI161 or TFPI-ΔK3. Deletion of the K3 domain did not affect the binding of FXa to TFPI (Figure 5C-D). Western blot analysis of TFPIWT and TFPI-ΔK3 using a rabbit polyclonal antibody against the C-terminus indicated that both TFPIWT and TFPI-ΔK3 contained an intact C-terminus (data not shown). This excluded the possibility that loss of binding to PS could have resulted from C-terminal degradation of the TFPI-ΔK3, which can occur when TFPI-α is purified from mammalian cell culture medium.15

Ligand blotting of PS and FXa to TFPI. TFPI proteins were electrophoresed on 12% SDS-PAGE gel and transferred to a nitrocellulose membrane. The amount of each protein that was electrophoresed and blotted is labeled at the top. (A-B) Binding of PS to TFPI. PS was used to probe the membrane, and the bound PS was detected by an anti-PS polyclonal antibody as described in “Ligand and Western blotting.” (C-D) Binding of FXa to TFPI. After PS detection in panels A and B, the membranes were stripped with 0.1mM glycine, pH 2.2, and reprobed with FXa. The bound FXa was then detected using a goat anti-FX polyclonal antibody as described in “Ligand and Western blotting.” The positions of molecular weight markers are indicated on the left.
Ligand blotting of PS and FXa to TFPI. TFPI proteins were electrophoresed on 12% SDS-PAGE gel and transferred to a nitrocellulose membrane. The amount of each protein that was electrophoresed and blotted is labeled at the top. (A-B) Binding of PS to TFPI. PS was used to probe the membrane, and the bound PS was detected by an anti-PS polyclonal antibody as described in “Ligand and Western blotting.” (C-D) Binding of FXa to TFPI. After PS detection in panels A and B, the membranes were stripped with 0.1mM glycine, pH 2.2, and reprobed with FXa. The bound FXa was then detected using a goat anti-FX polyclonal antibody as described in “Ligand and Western blotting.” The positions of molecular weight markers are indicated on the left.
Effect of PS and TFPI on the prothrombinase complex
TFPI-α has been shown to be a potent inhibitor of FXa in the prothrombinase complex in the absence of prothrombin.22 In the presence of plasma levels of prothrombin, however, thrombin generation by prothrombinase is not affected by physiologic concentrations of TFPI-α.27 As PS significantly enhances FXa inhibition by TFPI-α, we tested whether TFPI-α would inhibit prothrombin activation by prothrombinase in the presence of PS. When prothrombin was activated with the prothrombinase complex in the presence or absence of TFPI with or without PS at physiologic concentrations, a similar thrombin generation rate (∼ 2300nM/minute per nM) was obtained with TFPIWT and TFPI-ΔK3.
Effect of PS on FXa-induced coagulation
To assess whether deletion of the K3 domain and the loss of an interaction with PS affect TFPI inhibition of FXa activity in plasma, TFPIWT, TFPIK3P1, or TFPI-ΔK3 was added to plasma, and clotting times were recorded after the addition of FXa. The anticoagulant activity of TFPIK3P1 and TFPI-ΔK3 was similar to that of TFPIWT in PS-deficient plasma (Figure 6A). On the other hand, when pooled normal plasma or PS-deficient plasma reconstituted with PS was used, the activity of TFPIWT was substantially greater than that of TFPIK3P1 > TFPI-ΔK3 (Figure 6B-C).

Comparison of TFPIWT, TFPI-ΔK3, and TFPIK3P1 in FXa-induced coagulation of plasma. Assays were performed as described.27 Briefly, a 50/50 mixture of TFPI and plasma was incubated at 37°C for 3 minutes before adding a second mixture containing FXa, CaCl2, and cephalin, also preincubated at 37°C for 3 minutes, and the clotting time recorded using a fibrometer. Final concentrations for each reagent were TFPI (0-20nM), FXa (2nM), cephalin (25μM), and CaCl2 (5mM). Assays were performed in duplicate. (A) Assay with PS-deficient plasma (B) Assay with pooled normal plasma. (C) Assay with PS-deficient plasma spiked with 200nM PS. ● indicates TFPIWT; □, TFPIK3P1; and ▵, TFPI-ΔK3.
Comparison of TFPIWT, TFPI-ΔK3, and TFPIK3P1 in FXa-induced coagulation of plasma. Assays were performed as described.27 Briefly, a 50/50 mixture of TFPI and plasma was incubated at 37°C for 3 minutes before adding a second mixture containing FXa, CaCl2, and cephalin, also preincubated at 37°C for 3 minutes, and the clotting time recorded using a fibrometer. Final concentrations for each reagent were TFPI (0-20nM), FXa (2nM), cephalin (25μM), and CaCl2 (5mM). Assays were performed in duplicate. (A) Assay with PS-deficient plasma (B) Assay with pooled normal plasma. (C) Assay with PS-deficient plasma spiked with 200nM PS. ● indicates TFPIWT; □, TFPIK3P1; and ▵, TFPI-ΔK3.
Discussion
It was reported several years ago that PS possessed activated protein C-independent anticoagulant activity, perhaps related to the binding of PS to FXa and FVa.28 This “direct” PS anticoagulant activity varied substantially between different PS preparations, however, and subsequent studies suggested it was related to the presence of PS multimers.29 Whether multimers of PS are present in plasma or are generated during PS purification is controversial.29-33 More recently, it was shown that PS enhances the inhibition of FXa by TFPI-α and that this action of PS is apparent with monomeric PS.12,13,34
TFPI-α regulates blood coagulation by directly inhibiting FXa through its K2 domain and, in a FXa-dependent fashion, inhibiting FVIIa/TF through its K1 domain. The current studies demonstrate that the K3 domain and, to a lesser extent, the C-terminus of TFPI-α are important for the Ca2+ and PL-dependent PS enhancement of TFPI-α-mediated FXa inhibition. Although it is conceivable that the presence of the K3 domain imparts a specific, favorable conformation of the TFPI-α molecule that is required for its binding to PS, the ligand-binding results with the isolated K3 domain (Figure 5B) and the fact that a single amino acid change in K3 substantially reduces the PS-dependent enhancement of FXa inhibition by TFPI-α (Figure 3) strongly suggest a direct interaction between the K3 domain and PS is involved. Moreover, the equivalent direct inhibition of FXa produced by TFPIWT and TFPI-ΔK3 indicates that a lack of response to PS by TFPI-ΔK3 is not the result of misfolding of the mutant protein or misalignment of the C-terminal peptide relative to the K2 domain that binds to FXa. Yegneswaran et al have recently shown that monomeric PS interacts with FXa in a Ca2+ and PL-dependent fashion.35 Thus, the most straightforward explanation for the potentiating effect of PS on FXa inhibition by TFPI-α in the presence of Ca2+ and PL is that an association between PS and TFPI-α serves to increase TFPI-α's interactions with PL and with FXa at PL surfaces.
Previous studies have demonstrated that plasma levels of full-length TFPI-α are reduced in persons with deficiencies of factor V, factor VIII, or PS and that immunoprecipitation with anti–factor V and anti-PS antibodies reduces the level of full-length TFPI-α in normal plasma by 80% and 60%, respectively.36,37 These results have led to the suggestion that the binding of TFPI-α to these other coagulation proteins in plasma may protect TFPI-α from proteolysis and clearance. The analyses of the PS–TFPI-α binding by SPR presented here and elsewhere37 are not straightforward but suggest a binding constant (dissociation constant > 1μM), which is substantially greater than the plasma concentration of the free PS (160nM) to which TFPI-α reportedly binds.37 Perhaps the SPR studies are confounded by aggregation of PS at the higher concentrations tested or, alternatively, the mechanism(s) for the association of TFPI-α with these proteins in plasma is more complex than simple 1:1 interactions. Based on the functional assays (Figure 3; Table 1), however, the interaction of PS with TFPI-α and FXa at the PL surface in the presence Ca2+ is clearly of high affinity (EC50 ∼ 8nM).
The endothelium is the major site of TFPI-α synthesis,6 and a fraction of the TFPI-α produced by endothelial cells remains on the cell membrane, apparently associated with a glycosyl phosphatidylinositol-anchored moiety(ies).19 Previous studies have shown that optimal binding of endogenously produced TFPI-α at the cell surface requires the K3 domain and the C-terminus of TFPI-α and that the binding of TFPIK3P1 is substantially reduced. These binding characteristics mimic those described here for the PS-dependent enhancement of FXa inhibition by TFPI-α, suggesting that PS, which is expressed by endothelial cells, may be involved in the interaction of TFPI-α with the cell surface. Ongoing studies are investigating this possibility.
TFPI-α is a potent inhibitor of FXa in the prothrombinase complex in the absence, but not the presence, of prothrombin,22,27 and the presence of PS does not affect this result (“Effect of PS and TFPI on the prothrombinase complex”). This suggests that the stimulating effect of PS on FXa inhibition by TFPI-α would be operative early in coagulation, before the formation of the prothrombinase complex, or at local sites where prothrombin has been consumed.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
This work was supported by the National Institutes of Health (grant HL77193; G.J.B.).
National Institutes of Health
Authorship
Contribution: M.N. designed and performed the research, analyzed the data, and wrote the manuscript; E.A.T. constructed and expressed the TFPI-ΔK3 mutant and the K3 domain of TFPI-α; G.J.B. designed research, analyzed and interpreted the data, and wrote the manuscript; and all coauthors read and reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: George J. Broze Jr, Division of Hematology, Washington University, Campus Box 8125, 660 S Euclid Ave, St Louis, MO 63110; e-mail: gbroze@dom.wustl.edu.

