Abstract
Although highly active antiretroviral therapy has enabled constant progress in reducing HIV-1 replication, in some patients who are “aviremic” during treatment, the problem of insufficient immune restoration remains, and this exposes them to the risk of immune deficiency–associated pathologies. Various mechanisms may combine and account for this impaired immunologic response to treatment. A first possible mechanism is immune activation, which may be because of residual HIV production, microbial translocation, co-infections, immunosenescence, or lymphopenia per se. A second mechanism is ongoing HIV replication. Finally, deficient thymus output, sex, and genetic polymorphism influencing apoptosis may impair immune reconstitution. In this review we will discuss the tools at our disposal to identify the various mechanisms at work in a given patient and the specific therapeutic strategies we could propose based on this etiologic diagnosis.
Introduction
Highly active antiretroviral therapy (HAART) is increasingly efficient at reducing HIV-1 load. Currently, even with salvage therapy, up to 90% of treated HIV-1–infected adults are “aviremic,” that is, have viral RNA plasma levels under the level of detection of commercially available tests.1 Consequently, most of these patients reconstitute their immunity under treatment. Yet, in some patients, CD4+ T-cell recovery is abnormally low despite their full virologic response to HAART. An impaired immunologic response is linked to increased risk of disease progression and death,2-6 but we currently have no efficient therapeutic strategy in these situations. It is therefore becoming more and more important to understand the pathophysiologic mechanisms responsible for this lack of immune reconstitution, to design assays capable of diagnosing these mechanisms, and to propose therapies adapted to each situation.
Immunologic responses to HAART: both quality and quantity matter
Peripheral CD4+ T-cell increase under treatment is a 3-phase process, whatever the regimen is (Figure 1). During the first 1 to 6 months of HAART, the average rate of reconstitution is of 20 to 30 cells/μL monthly.7-10 Bosch et al have estimated this first phase to last for exactly 10 weeks.11 Most of this initial reconstitution seems to be the consequence of a redistribution of memory CD4+ T cells12 from the lymphoid tissues toward the blood compartment. The decrease in immune activation induced by the inhibition of viral replication results in a down-regulation of adhesion molecules at the surface of CD4+ T cells, including vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1). As these adhesion molecules are responsible for the trapping of CD4+ T cells into the secondary lymphoid organs, their down-regulation provokes the release of these CD4+ T cells into the periphery.13 The second phase of CD4+ T-cell increase, of about 5 to 10 cells/μL monthly, lasts until the end of the second year of therapy, and the third phase, of about 2 to 5 cells/μL monthly, extends beyond this second year for at least 7 years.10,12,14-16 Various mechanisms account for the rise in mostly naive CD4+ T cells12,17 during these 2 phases: first, de novo production of T lymphocytes by the thymus; second, the homeostatic proliferation of the residual CD4+ T cells, and third, the extension of CD4+ T-cell half life, a mechanism responsible for sustaining the number of naive CD4+ T cells in older individuals in whom thymic production is impaired.18 From a qualitative point of view, these 3 mechanisms of reconstitution are not equivalent. A healthy individual harbors a panel of T cells able to recognize a large set of antigens, his T-cell repertoire. HIV-induced CD4+ T-cell loss results in the shrinkage of this repertoire. If CD4+ T-cell recovery under therapy is based on the production of new CD4+ T cells, this repertoire may be at least partially restored. In contrast, if the increase in CD4 count stems mainly from CD4+ T-cell proliferation and survival, the T-cell repertoire will remain truncated, even though the total number of CD4+ T cells is normalized (Figure 2).

Average CD4+ T-cell recovery under HAART. The increase in CD4+ T-cell count and the mechanisms responsible for this increase are represented at various time points.
Average CD4+ T-cell recovery under HAART. The increase in CD4+ T-cell count and the mechanisms responsible for this increase are represented at various time points.

T-cell repertoire reduction under HIV-1 infection, and reconstitution under HAART. The quality of the reconstitution is represented depending on the various mechanisms accounting for the increase in CD4+ T-cell count.
T-cell repertoire reduction under HIV-1 infection, and reconstitution under HAART. The quality of the reconstitution is represented depending on the various mechanisms accounting for the increase in CD4+ T-cell count.
What is an impaired CD4+ T-cell restoration on HAART? Unfortunately there is no consensus on the definition. Some authors have called nonimmunologic responders patients whose CD4+ T-cell count remained below a threshold (eg, 350 to 500 cells/μL) after a variable period of time of treatment (eg, 4 to 7 years). For instance, Kelley et al showed that 24% of individuals with a CD4+ T-cell count < 500 cells/μL at year 4 of HAART had evidence of a CD4+ T-cell count plateau between years 4 and 7.5.19 It seems preferable to take into account the pretherapeutic CD4+ T-cell count and to define nonimmunologic responders after their CD4+ T-cell slope. A reasonable definition could be a rise in CD4+ T-cell count lower than 100 cells/μL after 2 years of treatment, that is, less than half of the expected recovery.
Because of the thus far lack of consensus on this definition, and because of the variability of the patient populations studied, the estimation of the frequency of virologic responder only varied from 6% to 24%.3-5,12,14,19-23
Of note, not only CD4+ T-cell number and quality are impaired in HIV infection, but many functions of many immune cells may be impaired, and not fully recovered under HAART. For instance, plasmacytoid dendritic cell and natural killer cell activities have been reported to remain incompletely reconstituted after 1 year of effective antiretroviral therapy.24 Thus, by routinely monitoring only CD4+ T-cell count, we overlook not only CD4+ T-cell functionality but also the functionality of the other immune cells. The observation that in vivo immune response to immunization may remain impaired in treated patients with more than 450 CD4+ T cells/μL25 emphasizes this notion that CD4 count is an imperfect surrogate marker of immune restoration. The failure of IL-2 therapy to protect from disease progression despite a robust effect on CD4 counts26 is in keeping with this idea. This means that we will need to define better indicators, for instance, subpopulations of CD4+ T cells, expression of specific CD4+ T-cell surface antigens, in vitro functional assays, or in vivo tests to monitor immune reconstitution.
The many determinants of impaired immune reconstitution in virologic responders
The reason why the drastic reduction in viral load does not always result in the normalization of the CD4+ T-cell count may be multifactorial. It is of major importance to understand these various factors at work if we want to establish an etiologic diagnosis and propose an appropriate therapy. Two main mechanisms may result in a suboptimal rise in CD4 count: insufficient production of CD4+ T cells and excessive CD4+ T-cell destruction. Excessive CD4+ T-cell destruction may be the consequence of HIV pathogenesis, immune activation, and/or genetically determined increase in the programmed cell death of lymphocytes (Figure 3).

Thymic output
The level of de novo T-cell production is a determining factor for CD4+ T-cell recovery. Many studies have shown that poor CD4+ T-cell regeneration is linked to a low proportion of naive cells among CD4+ T lymphocytes.27-31 Smith et al have reported a link between the importance of the naive CD4+ T-cell gain during the first 4 weeks of therapy and the abundance of thymic tissue.32 Freshly matured T cells, also called recent thymus emigrants (RTEs), are characterized by their harboring a scar that is a consequence of the rearrangement of the genes coding for the T-cell receptor (TCR). This scar is an extra-chromosomic circular DNA that is a by-product of the rearrangement, called a TCR excision circle (TREC). TRECs have been therefore used as a surrogate marker of thymus output, although cell division may dilute their content. Some,27,33 but not all,31 groups have reported low levels of TRECs in T lymphocytes in subjects with low CD4+ T-cell response. This might be the consequence of an impaired bone marrow30 and/or thymic34 production. It could also account for the frequent observation that older individuals, in whom thymic activity has declined,35 are at higher risk for persistently reduced CD4+ T-cell count during HAART than younger persons.5,36,37
Another factor that may hinder thymocyte production is HIV-1 infection per se, as it impairs hematopoietic stem cell metabolism38 and thymic function.39 As some of this impairment may be partially irreversible, T-cell maturation may remain suboptimal in some patients even though the viral replication is inhibited.
In final support of the importance of thymus output, polymorphisms in genes coding for the cytokines IL-2 and IL-15 and the receptor for IL-15, involved in the maturation and development of T-cells, have been associated with nonimmunologic response to HAART.40
Persistent viral replication
In aviremic patients under HAART, some cells may still produce virions, so that ultra-sensitive techniques can detect HIV RNA in the blood41,42 and in tissues.43 The question thus arises as to whether these residual virions stem from reservoir cells that continuously release noninfecting particles (residual production) or from recently infected cells that produce infectious particles that permanently infect new cells (ongoing replication). Whereas most authors agree that residual production persists in some patients, the possibility of ongoing replication is controversial. Various observations argue for the existence of ongoing replication. First, some authors,41,43,44 but not all,45 have observed the diversification of HIV sequences in aviremic patients, a phenomenon consecutive to mutations occurring during reverse transcription and therefore necessitating viral life cycles. Second, extra-chromosomic HIV DNA, including circular DNA containing 2 “long terminal repeat” sequences (LTR), a putative marker of recent infection, has been evidenced in virologic responders.46 Third, Petitjean et al have shown that under in vitro activation, the production of virions by peripheral blood CD4+ T cells from virologic responders may be prevented by an integrase inhibitor.47 These results indicate that some of these cells had preintegrated forms of viral genome, that is, had been recently infected. Likewise, recent reports of an increase in the amount of 2 LTR forms of HIV DNA in the peripheral blood mononuclear cells (PBMCs) of aviremic patients under treatment intensification with integrase inhibitor48 are in line with the same hypothesis. On the other hand, some others who observed that treatment intensifications have not reduced residual HIV-1 viremia in virologic responders argue against the hypothesis of ongoing replication.49-51 Finally, the finding that monocytes from subjects aviremic under HAART may harbor HIV proviral DNA52 is an additional argument for ongoing replication because these cells transit very rapidly in the blood.
Thus, if ongoing replication occurs in some aviremic patients, in lymphoid organs for instance, the cytopathogenicity of the virus may still be at work, particularly if it uses CXCR4 as a coreceptor,31 and this could hamper the reconstitution of the CD4+ T-cell count. Moreover, Doitsh et al have recently shown that the HIV replicative cycle does not need to be complete to induce CD4+ T-cell death.53
Immune activation
Many authors have linked immune activation to impaired rise in peripheral blood CD4+ T-cell count under treatment.28-30,54-56 This link may have at least 2 causes. On one hand, as discussed above, activated CD4+ T cells may be trapped in secondary lymphoid organs. And on the other hand, most CD4+ T cells die by apoptosis once they have been stimulated. Various phenomena, listed in the following sections, may be responsible for immune activation in the course of HIV-1 disease (Figure 3).
Residual viral production.
Persistent production of viral particles by reservoir cells in aviremic treated patients even in absence of ongoing replication may induce an immune activation. This is because of the anti-HIV immune response and of the fact that various HIV components are able to stimulate the immune system. HIV RNA for instance induces immune cell activation via TLR7,57 the gp120 envelope through the CD4 receptor and the coreceptors58 and the accessory protein nef through lipid rafts.59 All the more so, any ongoing replication should induce immune activation. In line with this, HAART intensification has been reported to decrease immune activation in some patients.60
Age.
Older persons have a higher background of immune activation than younger ones.61 This might contribute, in addition to their reduced thymic output, to the link between age and suboptimal CD4+ T-cell progression. From a general point of view, immunosenescence might be a cause and a consequence of immune activation, leading to a vicious circle.
High level of CCR5-induced activation.
As the main HIV-1 coreceptor, C-C chemokine receptor type 5 (CCR5) is now recognized as a co-activation molecule at the surface of CD4+ T cells,62 it could be involved in immune activation and thereby in the rise in CD4 count. This could explain why the administration of a CCR5 antagonist resulted in a decrease in T-cell activation.63,64 Accordingly, a high efficiency of CCR5-induced activation, as a consequence of genetic polymorphism65 or of a high CD4+ T-cell surface CCR5 density (P.C. and J.R., unpublished data, November 2010) may favor the persistence of immune activation under HAART. In support of this notion, Ahuja's group has established a correlation between CD4+ T-cell recovery and the genetic polymorphism of the gene encoding CCR5, and the number of copies of the gene encoding one of the natural ligand of this coreceptor, C-C chemokine ligand 3-like 1 (CCL3L1).65 In this study, the authors observed that this polymorphism was predictive of the intensity of CD4 count increase in virologic responders. This raises the interesting hypothesis that CCR5, in addition to its virologic roles as an HIV-1 coreceptor, might play a role in immune reconstitution. This hypothesis is in line with the fact that CD4+ T-cell restoration is linked to CD4+ T-cell surface CCR5 density.66
Co-infections.
Another possible source of CD4+ T-cell stimulation comes from infectious agents other than HIV. Hepatitis C virus (HCV) co-infection is known to result in a higher level of immune activation.67 Most but not all authors have linked HCV infection with incomplete CD4+ T-cell regeneration.68-70 Likewise, cytomegalovirus has been identified as a cause of CD8+ T-cell activation in HIV-infected subjects under HAART.71 Immune activation might also explain why CD4+ T-cell restoration after initiation of HAART is impaired in subjects who are co-infected with Mycobacterium avium complex.72
Microbial translocation.
It is now established that HIV-1 destroys the CD4+ T cells highly expressing CCR5 that are abundantly present in the gut-associated lymphoid tissues (GALT) early in the infection and that this destruction is partly irreversible, even after years of HAART.73 One of the consequences of this destruction is that the immune barrier becomes leaky, leaving the way for microbia coming from the gut lumen to invade the organism, a phenomenon called microbial translocation. Microbial translocation has been linked to the proportion of CD8+ T cells overexpressing CD38, and IFNα concentration in the blood.73 Therefore, microbial translocation might be one of the driver of immune activation, and thereby of CD4+ T-cell loss. In keeping with this notion, high levels of microbial translocation during therapy have been linked to reduced increases in the CD4+ T-cell count.74
Treg.
Predisposition to immune activation.
Genetics may predispose individuals to a high background of immune activation that might be deleterious for immune response under HAART. In keeping with this model, polymorphisms in genes encoding the cytokines IL-6, tumor necrosis factor-α (TNF-α), and interferon-α (IFN-α)40 have been involved in HAART-induced CD4+ T-cell count gain.75
Other genetic factors
Genes involved in apoptosis.
Apoptosis is a form of cell death responsible for CD4+ T-cell loss during HIV infection. The intensity of spontaneous,27 Fas-induced,27 and HIV-induced54 ex vivo apoptosis of CD4+ T cells has been inversely correlated with CD4+ T-cell progression after HAART. Therefore, it is not surprising that polymorphisms in genes involved in apoptosis such as TRAIL,40 Bim,40 Fas,76 and FasL76 have been linked to the rise in CD4+ T-cell count through therapy.
Sex.
Men have been reported to be at higher risk of non-CD4+ T-cell response under HAART than women.28,37 The reasons for that are unclear. One explanation might be that thymic output is higher in females than in males.35 Another could be an antiapoptotic effect of female sex steroids on CD4+ T cells as observed for instance on neutrophils.77 Moreover, the level of expression of CCR5 at the surface of CD4+ T cells, linked to CD4+ T-cell recovery66 as mentioned in “Immune activation,” is lower in women than in men.78
Other genes.
Other causes
HAART.
Some antiretroviral agents such as AZT may be toxic for hematopoietic progenitor cells,81 and could thereby hamper CD4+ T-cell count reconstitution. Likewise, the combination of tenofovir with high doses of ddI has been involved in poor recovery of CD4+ T cells eventually induced by a deleterious effect of ddI on T-cell maturation and differentiation.82 On the contrary, a better immunologic response has initially been reported with protease inhibitor.83 Yet other studies have shown that CD4+ T-cell gain was independent of the antiretroviral regimen.28,84 Recently, the integrase inhibitor raltegravir has been shown to induce a greater increase in CD4 count in treatment-naive patients at week 48 but not at week 96 compared with efavirenz.85 The same result was reported at week 48 for the CCR5 antagonist maraviroc, which persisted at week 96.86
CD4+ T-cell nadir.
Low nadir CD4+ T-cell count36,54,87 and low pretherapeutic CD4 count37,88 have been identified as predictive factors of persistently reduced CD4+ T-cell count. Obviously, if the definition of immune response is based exclusively on the CD4 count reached, rather than on the CD4 slope, the baseline CD4 count will definitely influence the level of immune response. Moreover, low nadir CD4+ T-cell count and low pretherapeutic CD4 count may cause emergence of X4 strains,89 intense microbial translocation,55 development of co-infections, and immunosenescence, all factors hindering CD4+ T-cell regeneration as discussed previously.
HIV RNA plasma level.
CD4+ T-cell response has been reported to be low when the pretherapeutic viremia is low.28 The reasons for this correlation remain to be unveiled.
How to identify the causes of an impaired CD4+ T-cell recovery in a given individual?
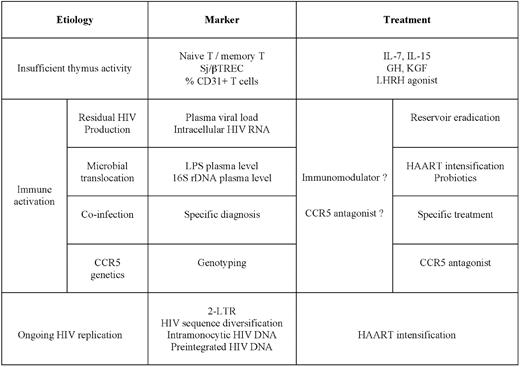
Now that we have reviewed the potential causes of poor CD4+ T-cell progression, failure of thymic activity, ongoing viral replication, and residual immune activation, it is important from a practical point of view to review the tools we have at our disposal to identify the main causes at work in a given patient (Table 1).
Insufficient thymic activity
A weak synthesis of new T cells will result in a low naive T-cell/memory T-cell ratio in peripheral blood. But, as other causes may have the same outcome, we need other tools to more specifically evaluate thymic activity. Various TRECs may be numerated, and the best indicator of thymic activity is the ratio between two of them, the signal-junction (sj)TREC and the βTREC, because this ratio is not modified by cell proliferation.39 However, determining the sj/βTREC ratio is not a routine assay, and we need more convenient markers. One of them could be CD31, as the CD31-expressing T-cell subpopulation contains the RTEs.90
Ongoing viral replication
We also need to design routine assays to identify the potential virologic responders in whom the virus still replicates. The relevance of the 2-LTR HIV DNA circles as a marker of recent infection is controversial, because the duration of their lifespan is discussed.91 The analysis of molecular variance of HIV sequences in patients who are aviremic under HAART as an indicator of continuous reinfection is protracted and requires a follow-up.44 The study of the effect of an integrase inhibitor on HIV-1 production induced by ex vivo stimulation of CD4+ T cells from patients under HAART to evidence the presence of unintegrated HIV DNA, a marker of recent infection, is also not a routine assay.47 Other strategies need to be tested, such as, for instance, the detection of HIV DNA in peripheral blood monocytes.52
Immune activation
The marker of immune activation that is the best predictor of disease progression might be the percentage of CD8+ T cells that overexpress CD38.92 But we also need to discriminate between the different causes of immune activation. Residual production of virions may be detected by quantitating the viremia with ultrasensitive techniques.41 Alternatively, the quantification of unspliced HIV RNA in PBMCs might be used to track persistent synthesis of viral particles.93 Active co-infection may be diagnosed by assays that are specific for the various infectious agents participating in the residual immune activation. High levels of CCR5 activation could be detected by genotyping the CCR5 gene and by determining the CCL3L1 gene copy number65 or by measuring CD4+ T-cell surface CCR5 density.66 Finally, microbial translocation has been initially measured by quantifying lipopolysaccharide (LPS) plasma level,94 but the quantification of bacterial ribosomal 16S DNA (16S rDNA) plasma level is more sensitive, more specific, and more comprehensive.74
Therapeutic possibilities
According to the etiology, various therapies may be considered (Table 1).
Insufficient thymic activity
Several options for increasing CD4+ T-cell restoration might be developed in the near future.
IL-7 is a good candidate because it promotes the maturation of thymocytes, the survival and function of T cells, as well as the homeostatic expansion of all T-cell subpopulations.95 A phase 1/2a trial has recently shown that administration of IL-7 enhanced T-cell recovery in HAART-treated subjects.96 The limitations might be that lymphopenic patients already produce high amounts of IL-7, and a potential drawback is that by inducing CXCR4 expression, IL-7 might favor the emergence of X4 strains.97 IL-15 has also been considered to boost immune recovery.98
Another option might be to inhibit the production of sex steroids. As estrogens, progesterone, and testosterone induce thymic atrophy, probably mainly through their effect on thymic stroma, castration has been tested with success to boost thymic activity in animals.99 Accordingly, in humans the administration of luteinizing hormone-releasing hormone (LHRH) agonist has resulted in transient enhancement of thymic function,100 particularly after hematopoietic stem cell transplantation.101 Obviously, although chemical castration is temporary, the gain in thymic output will have to be balanced with the side effects.
An alternative way to sustain thymic activity is to induce thymic epithelial cell proliferation by using keratinocyte growth factor (KGF). In mice, KGF administration reverses age-induced thymic and T-cell dysfunction.102 The effect of KGF after hematopoietic stem cell transplantation is currently being tested.
Finally, growth hormone (GH) is supposed to enhance hematopoiesis and immune function both directly and via insulin-like growth factor 1.103 GH treatment has been shown to improve thymic function in HIV-1–infected subjects.104 Yet, here again the risk of adverse events, such as carpal tunnel syndrome, hyperglycemia, or cancer progression, will have to be carefully considered.
Ongoing viral replication
If there is evidence for continuous reinfection of target cells in tissues, the challenge will be to intensify antiretroviral therapy with the aim of stopping this process. Attention should be paid to ensuring that the chosen therapeutic agents reach inhibiting concentrations in the organs where this replication persists.
Immune activation
Two strategies may be adopted: on one hand, a symptomatic therapy aimed at reducing the global level of polyclonal immune activation, and on the other hand, etiologic therapies targeting the mechanisms responsible for this activation.
Symptomatic therapy.
In the past, various attempts to down-regulate the polyclonal immune activation observed in HIV-1–infected patients have been made. These attempts included the use of hydroxyurea, thalidomide, mycophenolate, corticosteroids, IL-10, anti–TNF-α agents, cyclosporin A, FK506, and rapamycin.105 Most of these attempts have been disappointing. If CCR5 is actually involved, as a co-activation molecule, into this immune activation, CCR5 antagonists could exert some inhibitory effect on it. Of note, HAART intensification with the CCR5 antagonist maraviroc has resulted in a decrease in the proportion of activated peripheral blood CD4+64 and CD8+63 T cells, and in an increase in CD4+ T-cell slopes that tended to achieve significance63 in nonimmunologic responders. Thus, agents targeting CCR5 could have an immunologic effect, sparing CD4+ T cells in addition to their antiviral effects.
Residual viral production.
In this case, the way to stop HIV-induced immune activation will be to get rid of the reservoir cells that continue to release virions even though these virions do not cause de novo infection. This goal is presently pursued by some groups with different approaches including the brief induction of viral overexpression with the aim of inducing a cytopathic effect. For this purpose, cytokines as IL-2 or IL-7, activation of protein kinase C, agents remodeling the chromatin, intravenous immunoglobulin injection, and microRNA manipulation have been proposed.100-108
Co-infections.
If active co-infections reinforce the global immune activation, the infectious agents responsible for these co-infections might be targeted. Thus, therapeutic clearance of HCV RNA in HIV/HCV co-infected adults has been shown by some authors to decrease immune activation67 and to improve immune reconstitution under HAART.68 In the same way, 8 weeks of treatment with valganciclovir of CMV-seropositive HIV-infected patients with CD4+ T-cell counts < 350 cells/μL after more than 1 year of HAART resulted in a decrease in the proportion of activated CD8+ T cells.109
Microbial translocation.
The treatment of microbial translocation may be considered in at least 2 ways. First, if a persisting viral replication in the GALT prevents the reconstitution of the gut-associated immune barrier, intensification of HAART using a regimen of drugs with an efficient gut tissue penetration may be a solution. Second, therapeutic intervention, such as per os administration of probiotics, aimed at modifying the commensal microbiota to reduce the presence of proinflammatory bacteria and to increase that of anti-inflammatory bacteria,110 might be considered.
High level of CCR5 activation.
If co-activation signals delivered via CCR5 participate in the maintenance of a high level of immune activation, the administration of CCR5 antagonists might be beneficial.
Finally, the fact that some of the causes of incomplete immune response we have reviewed might be prevented by HAART argues for an early initiation of antiretroviral therapy.
Conclusions
Various mechanisms may be responsible in a given patient for an impaired immune recovery under HAART despite a control of viral replication. To correctly address this issue, we need to design convenient tools to identify which of these mechanisms are at work in each nonimmunologic responder. On the basis of this personalized etiologic diagnosis we then will be able to propose specific therapeutic strategies. Beyond the challenge of restoring immunity to prevent AIDS-associated events, the measurement of immune hyperactivation, the identification of the causes of this hyperactivation, and the fight against it will also decrease the risk of non–AIDS-linked pathologies.
Authorship
Contribution: P.C. wrote the manuscript; and J.R. reviewed and edited the manuscript.
Conflict-of-interest disclosure: P.C. has received honoraria for lectures or advisory boards from Bristol-Myers Squibb, GlaxoSmithKline, Merck Sharp & Dohme-Chibret, Pfizer, Gilead, and ViiV Healthcare, and a research grant from Pfizer. J.R. has received honoraria for lectures or advisory boards and/or research support from Abbott, Bristol-Myers Squibb, Boehringer Ingelheim, Gilead, GlaxoSmithKline, Merck Sharp & Dohme-Chibret, Pfizer, Roche, Schering-Plough, and Tibotec.
Correspondence: Pierre Corbeau, Laboratoire d'Immunologie, Hôpital Saint Eloi, 80 avenue A. Fliche, 34295, Montpellier cedex 5, France; e-mail: p-corbeau@chu-montpellier.fr.