

Abstract
Abstract 2574
Recently, using genome-wide single nucleotide polymorphism arrays and gene candidate deep exon sequencing, we identified lesions in CDKN2A gene, encoding p16/INK4A and p14/ARF tumor suppressors, in 27% (32/117) adult newly diagnosed Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL) patients and in 47% (14/30) relapsed cases. Clinically, in our cohort CDKN2A deletions were associated by univariate analysis to a worse outcome in terms of overall survival (OS), disease-free survival (DFS) and cumulative incidence of relapse (CIR) (OS: 27.7 vs 38.2 months, p = 0.0206; DFS: 10.1 vs. 56.1 months, p = 0.0010; CIR: 73.3 vs 38.1, p = 0.0014). Noteworthy, the negative prognostic impact of CDKN2A deletion on DFS was also confirmed by the multivariate analysis (p = 0.0051). These results showed that there are genetically distinct Ph+ ALL patients with a different risk of leukemia relapse and that testing for CDKN2A alterations at diagnosis may help in risk stratification. Furthermore, since the loss of CDKN2A eliminates the critical tumor surveillance mechanism and allows proliferation and tumor cell growth by the action of MDM2, a negative regulator of p53, we investigated the preclinical activity of the MDM2 antagonist RG7112 in primary B-ALL patient samples and leukemic cell line models. BV-173, SUPB-15 and K562 Ph+ cell lines were incubated with increasing concentration of RG7112 (0.5–10 μM) and its inactive enantiomer for 24, 48 and 72 hours (hrs). MDM2 inhibition by RG7112 resulted in a dose and time-dependent cytotoxicity with IC50 (24 hrs) of 2 μM for BV-173 and SUPB-15 which harbor homozygous deletion of CDKN2A but wild-type p53. No significant changes in cell viability were observed in K562 p53-null cell line after incubation with RG7112. The time and dose-dependent reduction in cell viability were confirmed in primary blast cells from a Ph+ ALL patient with the T315I Bcr-Abl kinase domain mutation found to be insensitive to the available tyrosine kinase inhibitors and from a t(4;11)-positive ALL patient (IC50 at 24 hrs equal to 2 μM). Consistent with the results of cell viability, Annexin V/Propidium Iodide analysis showed a significant increase in apoptosis after 24 hrs in BV-173, SUPB-15 and in primary leukemia blasts, whereas no apoptosis was observed in K562 cells. To examine the possible mechanisms underlying RG7112-mediated cell death, western blot analysis was performed. Protein levels of p53, p21 (an important mediator of p53-dependent cell cycle arrest), cleaved caspase-3 and caspase-9 proteins increased upon treatment with RG7112 after 24 hrs of incubation with concentrations equal to the IC50. These data demonstrate the ability of RG7112 to activate the intrinsic apoptotic pathway by a p53-dependent mechanism. In order to better elucidate the implications of p53 activation and to identify biomarkers of clinical activity, gene expression profiling analysis (Affymetrix GeneChip Human Gene 1.0 ST) was next performed, comparing sensitive cell lines (BV-173 and SUPB-15) after 24 hrs exposure to 2 μM RG7112 and their untreated counterparts (DMSO 0.1%). A total of 621 genes (48% down-regulated vs 52% up-regulated) were differentially expressed (p < 0.05). They include genes involved in cell cycle and apoptosis control (e.g. Histone H1, TOP2, GAS41, H2AFZ) and in the down-regulation of the Hedgehog signaling (e.g. BMI1, BMP7, CDKN1C, POU3F1, CTNNB1, PTCH2) with a strong repression of stemness genes and re-activation of INK4/ARF as illustrated in Figure 1. Actually, both GAS41 (growth-arrest specific 1 gene) and BMI1 (a polycomb ring-finger oncogene) are repressors of INK4/ARF and p21 and their aberrant expression has found to contribute to stem cell state in tumor cells. In our data they were strongly down-regulated (fold-change −1.35 and −1.11, respectively; p-value 0.02 and 0.03, respectively) after in vitro treatment as compared to control cells, suggesting that these genes have a potential as new biomarkers of activity. In conclusion, inhibition of the p53–MDM2 interaction by RG7112 can activate the p53 pathway, resulting in apoptosis and inhibition of stemness genes in B-ALL with wild-type p53. Our findings provide a strong rational for further clinical investigation of RG7112 in Ph+ ALL. Supported by: ELN, AIL, AIRC, Fondazione Del Monte di Bologna e Ravenna, FIRB 2006, Ateneo RFO grants, Project of integrated program, Programma di Ricerca Regione–Università 2007–2009.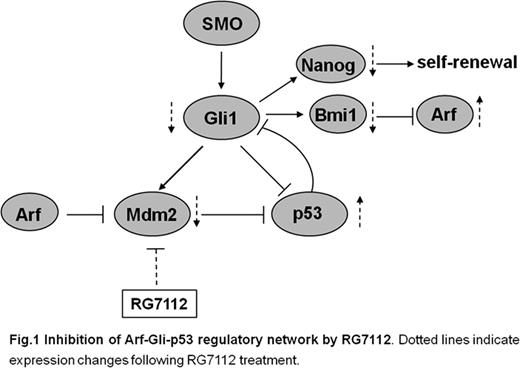

Disclosures:
Baccarani:Novartis: Consultancy; Bristol Myers Squibb: Consultancy; Novartis: Honoraria; Bristol Myers Squibb: Honoraria; Pfizer: Honoraria; Ariad: Honoraria. Martinelli:Novartis: Consultancy, Honoraria; BMS: Consultancy, Honoraria; Pfizer: Consultancy.

