

Abstract
EP217609 is a new dual-action parenteral anticoagulant that combines an indirect factor Xa inhibitor (fondaparinux analog) and a direct thrombin inhibitor (α-NAPAP analog) in a single molecule together with a biotin tag to allow avidin neutralization. EP217609 exhibits an unprecedented pharmacologic profile in showing high bioavailability, long plasma half-life, and potent antithrombotic activity in animals without the complications of thrombin rebound. Here we report the exceptional specificity and selectivity profile of EP217609. EP217609 inhibited thrombin with rapid kinetics (kon > 107M−1s−1), a high affinity (KI = 30-40pM), and more than 1000-fold selectivity over other coagulation and fibrinolytic protease targets, comparing favorably with the best direct thrombin inhibitors known. EP217609 bound antithrombin with high affinity (KD = 30nM) and activated the serpin to rapidly (kass ∼ 106M−1s−1) and selectively (> 20-fold) inhibit factor Xa. The dual inhibitor moieties of EP217609 acted largely independently with only modest linkage effects of ligand occupancy of one inhibitor moiety on the potency of the other (∼ 5-fold). In contrast, avidin binding effectively neutralized the potency of both inhibitor moieties (20- to 100-fold). These findings demonstrate the superior anticoagulant efficacy and rapid avidin neutralizability of EP217609 compared with anticoagulants that target thrombin or factor Xa alone.
Introduction
Heparin has been the reference anticoagulant drug of choice for the prevention and treatment of venous thrombosis for the past 70 years.1 The anticoagulant activity of this natural glycosaminoglycan derives from a specific pentasaccharide sequence that binds and activates the plasma serpin inhibitor of blood coagulation proteases, antithrombin.2,3 However, numerous off-target interactions of heparin with other plasma proteins and cells compromise the effectiveness and safety of this anticoagulant. Such interactions result in variable dosage requirements and consequent risks of bleeding, the danger of eliciting the immune syndrome, heparin-induced thrombocytopenia, and problems with thrombin rebound after anticoagulant therapy.1,4-7 The heparin pentasaccharide mimetics, fondaparinux and idraparinux, were developed to eliminate such off-target interactions and have been shown to be specific antithrombin-mediated inhibitors of factor Xa with predictable pharmacokinetic profiles and no risk of heparin-induced thrombocytopenia.8-11 However, problems with thrombin–rebound remain with these mimetics, which appear to result from the inability to inhibit clot-bound thrombin.12
The predictable pharmacokinetics of fondaparinux was the impetus for developing a dual factor Xa and thrombin inhibitor that combines a fondaparinux-like antithrombin-mediated inhibitor of factor Xa with an α-NAPAP–like direct thrombin inhibitor.13-16 The first compound of this generation, EP42675 (Figure 1), inhibits free and clot-bound thrombin in a manner like that of the related direct reversible thrombin inhibitor and retains the anti–factor Xa activity, complete bioavailability, long half-life, and predictable pharmacokinetic properties of fondaparinux. Moreover, it shows a strong antithrombotic activity in both arterial and venous thrombosis models and no evidence of rethrombosis in an experimental thrombolysis model.17 Addition of a biotin tag to the structure of EP42675 yielded EP217609 (Figure 1) that retained the anti-IIa and anti–factor Xa activities and pharmacokinetics of EP42675 and could be rapidly neutralized by avidin injection.15 In humans, administration of avidin triggered a rapid and irreversible neutralization of EP217609 without rebound effect.18

Structure of EP217609. The indirect factor Xa inhibitor moiety (fondaparinux analog), direct thrombin inhibitor moiety, and biotin label of EP217609 are indicated. The biotin is attached to the linker between the 2 inhibitor moieties through a lysine spacer indicated by the arrow. EP217609 is a mixture of diastereomers containing either an L-lysine spacer (EP217609-1) or a D-lysine spacer (EP307138-1). EP42675 lacks the lysyl-biotinyl group indicated by a box.
Structure of EP217609. The indirect factor Xa inhibitor moiety (fondaparinux analog), direct thrombin inhibitor moiety, and biotin label of EP217609 are indicated. The biotin is attached to the linker between the 2 inhibitor moieties through a lysine spacer indicated by the arrow. EP217609 is a mixture of diastereomers containing either an L-lysine spacer (EP217609-1) or a D-lysine spacer (EP307138-1). EP42675 lacks the lysyl-biotinyl group indicated by a box.
EP217609 is currently under investigation in a phase 2 clinical trial as a reversible anticoagulant in cardiopulmonary bypass (CPB) surgery. Surprisingly, the current standard of care for anticoagulation in CPB remains heparin and its neutralizing agent protamine, as safer or more efficacious anticoagulants have yet to be found.19 Low molecular weight heparin or the heparin-like danaparoid has been used in CPB, but they produce inadequate thrombin inhibition and are not neutralized by protamine.20,21 The direct thrombin inhibitors bivalirudin and argatroban have also been tested in patients, but none of these agents represents a real alternative to the heparin-protamine couple.22,23 A new approach based on the use of a reversible selective factor IXa inhibitor is promising but has not been tested in humans.24
Although EP217609 shows a promising pharmacologic profile, quantitative biochemical studies of the potency and selectivity of the dual inhibitor for its targets have not been done to provide a biochemical foundation for understanding its exceptional pharmacologic efficacy. Here we demonstrate that EP217609 is among the highest affinity (KI = 30-40pM) and selective (> 1000-fold) direct thrombin inhibitors known and is a rapid (kass ∼ 106M−1s−1) and selective (> 20-fold) antithrombin-mediated inhibitor of factor Xa. Avidin binding directly neutralizes the dual inhibitor activities in addition to the indirect neutralization that results from accelerated clearance of the drug. Our findings support the promise of EP217609 as a next-generation anticoagulant capable of reversible inhibition of both existing and newly generated thrombin. Current phase 2 clinical trials evaluating EP217609 anticoagulant efficacy in CPB will provide an important test of this promise.
Methods
Materials
Proteases of human origin were from Enzyme Research Laboratories, and their concentrations determined by active-site titrations, as in prior studies.25 Human antithrombin was purified from blood plasma as described.26 Concentrations of the protein were determined from the absorbance at 280 nm using an extinction coefficient of 37 700M−1cm−1.27 The recombinant extracellular domain of human tissue factor was a gift of the late Dr Yale Nemerson. Egg white avidin was provided by Endotis Pharma (EP). The concentration of avidin solutions was determined by HPLC comparing the area under the curve with that of a standard of known concentration. All proteins were judged pure by SDS-PAGE analysis.28 Synthetic p-nitroanilide chromogenic and 7-amido-4-methylcoumarin (AMC) fluorogenic protease substrates were purchased from Chromogenix, American Diagnostica, Centerchem, or Sigma-Aldrich. Substrate concentrations were determined by absorbance using published extinction coefficients.25 The heparin pentasaccharide mimetics, fondaparinux and idraparinux,8,9 were provided by Sanofi-Aventis, and their concentration was determined by weight from the molecular weight. EP compounds were provided as aqueous solutions whose molar concentrations were determined by nuclear magnetic resonance spectroscopy using an internal standard.
Experimental conditions
All experiments, except where otherwise noted, were performed at 25°C in an I 0.15 buffer consisting of 100mM HEPES, 93.5mM NaCl, 5mM CaCl2, 0.1% PEG 8000, pH 7.4.
Binding of EP compounds to antithrombin and thrombin
Stoichiometries and dissociation constants for the binding of EP compounds to antithrombin were measured by fluorescence titrations as in past studies.26 Stepwise additions of compounds to antithrombin were made followed by monitoring the increases in protein fluorescence that signal binding of the heparin pentasaccharide mimetic group using λex 280 nm and λem 340 nm. Titrations to determine binding stoichiometry were done in low ionic strength buffer consisting of 20mM sodium phosphate, 0.1mM EDTA, 0.1% PEG 8000, pH 7.4, whereas titrations to assess binding affinity were done in the I 0.15 HEPES buffer used for all other experiments. Fluorescence changes were fit by the quadratic equilibium binding equation.26 For stoichiometric titrations, KD was fixed based on the ionic strength dependence of KD measured for fondaparinux,10 and the maximal fluorescence change and the stoichiometric factor were fit as adjustable parameters. The fitted stoichiometric factor was assumed in titrations to determine binding affinity, in which case KD and the maximal fluorescence change were the fitted parameters.
Binding of the EP compounds to thrombin was measured by fluorescence titrations in the presence of the probe, p-aminobenzamidine,26 by following the decrease in fluorescence at 370 nm (λex 330 nm) caused by the displacement of bound p-aminobenzamidine from the thrombin active site that accompanies compound binding. Fluorescence changes were fit by the quadratic equilibrium binding equation. The KD for the thrombin–EP compound interaction in these fits was fixed at the measured KI value.26 The maximal fluorescence change and stoichiometric factor were the fitted parameters.
Measurements of KI values for protease inhibition by the EP compounds
EP compounds were incubated with protease for several minutes followed by the addition of chromogenic or fluorogenic substrates to yield final concentrations of 10−10 to 10−4M EP compound, 0.1 to 5nM protease, and 50 to 100μM substrate. The initial rate of substrate hydrolysis was then monitored continuously from the linear rate of absorbance change at 405 nm or fluorescence change at λex 380 nm, λem 440 nm. For experiments with factor IXa, hirudin was included at 30nM in reaction mixtures to inhibit traces of thrombin (estimated at 0.6% of factor IXa) because of the poor reactivity of factor IXa with the synthetic substrate under the experimental assay conditions. The dissociation constant for inhibitor binding, KI, was evaluated for all proteases except thrombin by fitting the decrease in the observed initial velocity of substrate hydrolysis, vobs, as a function of EP inhibitor concentration, [I]o, by the hyperbolic equation,
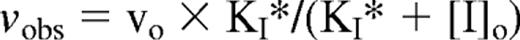
where vo is the initial velocity of substrate hydrolysis in the absence of inhibitor and KI* is the apparent dissociation constant for inhibitor binding.29 KI* is related to KI by a factor that accounts for the competitive effect of substrate binding:
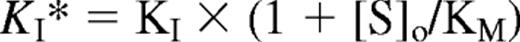
where [S]o is the fixed substrate concentration used and KM is the Michaelis constant for the protease-substrate interaction. Because of the tight binding of the EP compounds to thrombin, the decrease in observed initial velocities of substrate hydrolysis with increasing EP compound concentration was fit using the quadratic equation:
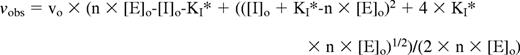
where [E]o is the total protease concentration, n is a stoichiometric factor, and all other parameters are as indicated in the hyperbolic equation above.29 The fitted parameters were vo, n, and KI*. It should be noted that, under conditions where inhibitor dissociation from the protease is slow relative to the time over which substrate hydrolysis is measured, the substrate will only interact with the free protease and not compete with the inhibitor-bound protease and therefore KI* will be equal to KI.
Kinetics of protease reactions with antithrombin–EP compound complexes
Kinetic studies of the reactions of antithrombin–EP compound complexes or antithrombin–idraparinux complex with proteases were performed similar to previous studies.25 Idraparinux was chosen as a reference because of its high affinity for antithrombin and because it activates antithrombin identical to fondaparinux.25 EP compound or idraparinux concentrations were in 1.5-fold molar excess over the antithrombin concentration, except for reactions of proteases that bound the α-NAPAP–like unit of the EP compounds with submicromolar KI values in which case the EP compound was equimolar with or lower than the antithrombin concentration to minimize competitive inhibition by the free EP compound. In all cases, the protease concentration was at least 10-fold lower than the concentration of antithrombin–EP compound complex to ensure pseudo-first-order reaction conditions. Reactions were initiated by adding protease to antithrombin and EP compound or idraparinux followed by quenching after different time intervals with chromogenic or fluorogenic substrate solution.25 Factor Xa reactions were performed with a fixed antithrombin concentration and varying EP compound or idraparinux concentrations for a fixed time of 300 seconds and then quenched with substrate. Substrate quench solutions for factor IXa reactions contained ethylene glycol to enhance the reactivity of the enzyme with the substrate and preclude any interference from traces of thrombin.30 The residual enzyme activity was measured by monitoring the initial linear increase in absorbance or fluorescence for several minutes. Progress curves of the loss of enzyme activity with time were fit by a single exponential decay function to obtain the observed pseudo-first-order inhibition rate constant (kobs).25 Second-order rate constants (kass) were calculated by dividing kobs by the concentration of antithrombin–EP compound complex calculated from measured KD values for the interactions. The contribution of the free antithrombin reaction with protease was negligible relative to the faster reaction of the antithrombin–EP compound complex. At least 2 inhibitor-compound complex concentrations were analyzed, and calculated second-order rate constants were averaged. Errors reported represent the range of observed values for 2 to 5 independent experiments. For factor Xa reactions, the loss of enzyme activity as a function of EP compound concentration was fit by a single exponential and the second-order association rate constant calculated by dividing the fitted exponential rate constant by the fixed reaction time and then correcting for the fraction of EP compound complexed with antithrombin based on the measured KD.31
Linkage effects
The effect of antithrombin binding to the EP compound on the KI for thrombin binding was analyzed according to the linkage scheme and equation:
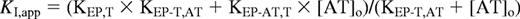
where KI,app is the apparent KI measured in the absence or presence of antithrombin, [AT]o is the total antithrombin concentration, and KEP,T, KEP-T,AT, and KEP-AT,T are dissociation constants for thrombin binding to free EP, for antithrombin binding to the binary EP-thrombin complex, and for thrombin binding to the binary EP–antithrombin complex, respectively. KEP-T,AT was determined by comparing the kinetics of factor Xa inhibition by antithrombin complexes with free EP versus EP-thrombin binary complex based on the equation:
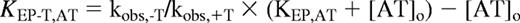
where kobs,-T/kobs,+T is the ratio of observed pseudo-first-order rate constants for reactions of antithrombin complexes with free EP and EP-thrombin binary complex and KEP,AT is the dissociation constant for antithrombin binding to free EP. The measured values of KI,app, KEP,T, and KEP-T,AT, were then used to calculate KEP-AT,T from the linkage equation.
Results
EP compounds bind tightly to antithrombin and thrombin
The ability of the EP compounds to bind both antithrombin and thrombin with high affinity was initially assessed by equilibrium binding titrations. Titrations of antithrombin with EP42675 and its biotin conjugate, EP217609 (Figure 1), were monitored by changes in the tryptophan fluorescence of antithrombin that report binding of the fondaparinux-like moiety of the compounds.10 At low ionic strength, the EP compounds produced identical linear increases in fluorescence up to an endpoint corresponding to an ∼ 40% fluorescence enhancement after ∼ 1 mol of compound/mol antithrombin had been added (Figure 2A), indicating stoichiometric binding of the compounds to the serpin. Titrations at physiologic ionic strength resulted in more gradual hyperbolic increases in fluorescence with increasing additions of the 2 compounds that indistinguishably approached saturable endpoints equivalent to those observed at low ionic strength. Fitting of the latter titrations by the equilibrium binding equation indicated identical KD values of 30 to 32nM, representing binding affinities ∼ 2-fold stronger than fondaparinux (KD = 60nM), but ∼ 60-fold weaker than the mimetic, idraparinux (KD = 0.4nM), which has an additional 2 sulfates9,32 (Table 1).

Equilibrium binding of antithrombin and thrombin to EP compounds. (A) Relative protein fluorescence changes (Fobs/Fo) accompanying the stepwise addition of EP42675 (open symbols) or EP217609 (closed symbols) to 500nM antithrombin in I 0.05 sodium phosphate buffer (○, ●) or to 200nM antithrombin in I 0.15 HEPES plus Ca2+ buffer (▵, ▴) both at pH 7.4, 25°C, are plotted as a function of the molar ratio of EP compound to antithrombin. Solid lines indicate fits to the quadratic equilibrium binding equation from which the binding stoichiometry, the KD (fixed at 0.5nM for I 0.05 titrations) and the maximal fluorescence change were derived. (B) Relative changes in p-aminobenzamidine fluorescence (Fobs/Fo) accompanying the stepwise addition of EP42675 (○) or EP217609 (●) to 200nM thrombin plus 100μM p-aminobenzamidine are plotted as a function of the molar ratio of EP compound to thrombin. Titrations were fit by the quadratic equilibrium binding equation in which KD was fixed at a value of 0.1nM based on measurements of KI and the competitive effect of probe binding to obtain values for the binding stoichiometry and maximal fluorescence change.
Equilibrium binding of antithrombin and thrombin to EP compounds. (A) Relative protein fluorescence changes (Fobs/Fo) accompanying the stepwise addition of EP42675 (open symbols) or EP217609 (closed symbols) to 500nM antithrombin in I 0.05 sodium phosphate buffer (○, ●) or to 200nM antithrombin in I 0.15 HEPES plus Ca2+ buffer (▵, ▴) both at pH 7.4, 25°C, are plotted as a function of the molar ratio of EP compound to antithrombin. Solid lines indicate fits to the quadratic equilibrium binding equation from which the binding stoichiometry, the KD (fixed at 0.5nM for I 0.05 titrations) and the maximal fluorescence change were derived. (B) Relative changes in p-aminobenzamidine fluorescence (Fobs/Fo) accompanying the stepwise addition of EP42675 (○) or EP217609 (●) to 200nM thrombin plus 100μM p-aminobenzamidine are plotted as a function of the molar ratio of EP compound to thrombin. Titrations were fit by the quadratic equilibrium binding equation in which KD was fixed at a value of 0.1nM based on measurements of KI and the competitive effect of probe binding to obtain values for the binding stoichiometry and maximal fluorescence change.
Figure 2B shows equilibrium binding titrations of thrombin with the EP compounds monitored from decreases in fluorescence of the probe, p-aminobenzamidine, that report the competitive displacement of the probe from the enzyme active site as the compounds bind.33 Titrations of thrombin with both EP compounds showed equivalent linear decreases in probe fluorescence to an endpoint fluorescence corresponding to that of the free probe after ∼ 1 mol of compound/mol thrombin had been added. This indicated tight stoichiometric binding of the EP compounds to the active site of thrombin.
The endpoints observed in the stoichiometric titrations of thrombin and antithrombin with the EP compounds were experimentally indistinguishable and showed equivalent minor deviations (∼ 30%) from theoretical 1:1 endpoints, presumably because of errors in compound concentrations determined by nuclear magnetic resonance. For consistency, corrected compound concentrations, obtained by equating concentrations observed at the titration endpoints with the antithrombin or thrombin concentrations used, were used in measuring KI values for direct inhibition of proteases and in determining second-order association rate constants for antithrombin-dependent inhibition of proteases.
EP compounds are highly selective active-site-directed inhibitors of thrombin
The relative inhibitory potency of the α-NAPAP–like moiety of the EP compounds against 7 procoagulant proteases and 3 anticoagulant proteases was evaluated by equilibrium binding titrations under conditions approximating physiologic (I 0.15, pH 7.4, 5mM Ca2+), although at a temperature of 25°C. Titrations were monitored from the inhibitory effect of the compounds on the initial velocity of hydrolysis of specific chromogenic or fluorogenic substrates by the proteases (Figure 3).

Specificity and selectivity of EP compounds as direct inhibitors of blood coagulation proteases. The relative decrease of the initial velocity of hydrolysis of specific reporter substrates by the indicated proteases is shown as a function of EP compound concentration. Data for the 6 proteases that were significantly inhibited up to 10−4M EP compound are shown as a function of increasing EP42675 (open symbols) or EP217609 (solid symbols) concentration. Data for EP217609 only are presented for the 4 proteases whose activity was insignificantly inhibited over the same EP compound concentration range. Solid and dashed lines indicate fits of EP217609 and EP42675 data, respectively, by equations for tight binding or regular competitive inhibition, which yielded values of KI*. KI was calculated from KI* by dividing the latter by the factor for competitive substrate inhibition, except for the KI* measured for thrombin, which required no correction. KI values are tabulated in Table 2.
Specificity and selectivity of EP compounds as direct inhibitors of blood coagulation proteases. The relative decrease of the initial velocity of hydrolysis of specific reporter substrates by the indicated proteases is shown as a function of EP compound concentration. Data for the 6 proteases that were significantly inhibited up to 10−4M EP compound are shown as a function of increasing EP42675 (open symbols) or EP217609 (solid symbols) concentration. Data for EP217609 only are presented for the 4 proteases whose activity was insignificantly inhibited over the same EP compound concentration range. Solid and dashed lines indicate fits of EP217609 and EP42675 data, respectively, by equations for tight binding or regular competitive inhibition, which yielded values of KI*. KI was calculated from KI* by dividing the latter by the factor for competitive substrate inhibition, except for the KI* measured for thrombin, which required no correction. KI values are tabulated in Table 2.
Inhibition of thrombin enzymatic activity by the EP compounds was essentially stoichiometric when ∼ 1nM thrombin was titrated, indicating tight binding (Figure 4A). Excellent fits of the titrations by the equation for a tight binding competitive inhibitor were observed. Indistinguishable apparent KI values were found when thrombin titrations were performed in the presence of substrates with a low KM (S2238) or high KM (S2366; data not shown), indicating that the reporter substrates measure equivalent free thrombin concentrations in the titrations because of the slow dissociation of bound compound from thrombin during the time of measurement. The apparent KI thus represents the true KI and requires no correction for substrate binding.29
![Figure 4. Equilibrium and kinetics of EP compound binding to thrombin. (A) Comparison of equilibrium binding titrations of 0.12 and 1.2nM thrombin with EP217609 monitored from the decrease in the initial rate of S2366 substrate hydrolysis by thrombin. Solid lines indicate fits by the equation for tight binding competitive inhibition. (B) Plot of kobs versus the effective [EP compound], obtained by dividing by the factor, 1 + [S]o/KM, for the binding of EP42675 (○) and EP217609 (●) to thrombin. kobs was measured from exponential progress curves of the time-dependent inhibition of thrombin hydrolysis of the low KM substrate, tosyl-GPR-AMC. The slope of this plot provided kon and the intercept provided koff.](/view-large/figure/7233228/zh89991285190004.jpeg)
Equilibrium and kinetics of EP compound binding to thrombin. (A) Comparison of equilibrium binding titrations of 0.12 and 1.2nM thrombin with EP217609 monitored from the decrease in the initial rate of S2366 substrate hydrolysis by thrombin. Solid lines indicate fits by the equation for tight binding competitive inhibition. (B) Plot of kobs versus the effective [EP compound], obtained by dividing by the factor, 1 + [S]o/KM, for the binding of EP42675 (○) and EP217609 (●) to thrombin. kobs was measured from exponential progress curves of the time-dependent inhibition of thrombin hydrolysis of the low KM substrate, tosyl-GPR-AMC. The slope of this plot provided kon and the intercept provided koff.
Equilibrium and kinetics of EP compound binding to thrombin. (A) Comparison of equilibrium binding titrations of 0.12 and 1.2nM thrombin with EP217609 monitored from the decrease in the initial rate of S2366 substrate hydrolysis by thrombin. Solid lines indicate fits by the equation for tight binding competitive inhibition. (B) Plot of kobs versus the effective [EP compound], obtained by dividing by the factor, 1 + [S]o/KM, for the binding of EP42675 (○) and EP217609 (●) to thrombin. kobs was measured from exponential progress curves of the time-dependent inhibition of thrombin hydrolysis of the low KM substrate, tosyl-GPR-AMC. The slope of this plot provided kon and the intercept provided koff.
The measured KI for binding of the nonbiotinylated EP42675 to thrombin was 61 ± 20pM (Table 2). The biotin label of EP217609 increased the affinity to 43 ± 16pM, although the significance of these differences was marginal because of the large errors in KI at the protease concentration used (∼ 20 × KI). Titrations performed at a 10-fold lower thrombin concentration confirmed the KI values measured at higher thrombin concentration and the modestly higher affinity of thrombin for the biotin-labeled than the unlabeled compound (Figure 4A).
Continuous measurements of the kinetics of EP compound inhibition of thrombin were performed by adding thrombin to mixtures of the low KM fluorogenic substrate and EP compound and following the exponential decrease in the rate of substrate hydrolysis to a final equilibrium inhibited value under pseudo–first-order conditions. From measurements of kobs as a function of inhibitor concentration, identical values for kon of 3.6± 0.2 × 107M−1s−1 and similar values for koff of 1 to 5 ± 4 × 10−4 s−1 were determined for EP42675 and EP217609 from the observed linear relationship between kobs and EP concentration (Figure 4B). These kinetic constants were consistent with the measured KIs and confirmed an extremely slow rate of inhibitor dissociation from thrombin (t1/2 > 20 minutes).
The substrate hydrolysis activity of 5 other proteases was inhibited by the EP compounds over the range of inhibitor concentrations examined (Figure 3). Because inhibitor concentrations greatly exceeded protease concentrations in these titrations, inhibition of each of the proteases was well fit by the equation for simple hyperbolic competitive inhibition. Corrections of apparent inhibition constants for substrate binding were applied in each case. Two proteases, factor XIa and activated protein C (aPC), bound the EP compounds with moderately high affinities (Table 2). aPC bound the parent EP42675 with a KI of 40 ± 3nM, an affinity still 1000-fold weaker than that for thrombin. The biotin label modestly weakened the affinity to a KI of 50 ± 6nM. Factor XIa bound EP42675 with a KI of 290 ± 20nM, an affinity ∼ 5000-fold weaker than that for thrombin. The binding was significantly improved with the biotin-labeled EP217609 (KI = 170 ± 10nM). EP42675 bound the 3 proteases, plasmin, kallikrein, and factor Xa with KI values of 5.2 ± 0.3μM, 22 ± 1μM, and 82 ± 4μM, respectively, that reflected affinities more than 100 000-fold weaker than that for thrombin. The biotin-labeled EP217609 modestly enhanced binding to these proteases by 2- to 3-fold (Table 2). No evidence for any biphasic slow inhibition kinetics reflecting a possible 2-step inhibition mechanism was observed for any of the EP compound-protease interactions.29 The compounds thus appear to inhibit all proteases through a simple one-step reversible equilibrium.
No significant inhibition by the EP compounds was observed for several of the proteases up to 10−5M compound, including factor IXa, factor VIIa-tissue factor complex, factor XIIa, and tPA (Figure 3). KI values for EP compound binding to these proteases were thus estimated to greatly exceed 10μM (Table 2).
Antithrombin–EP compound complexes selectively inhibit factor Xa
Progress curves for the inhibition of the enzymatic activity of the procoagulant and anticoagulant proteases by antithrombin complexes with EP compounds and with the reference compound idraparinux were compared under the same conditions as KI determinations and under pseudo-first-order reaction conditions. Observed pseudo-first-order rate constants (kobs) for the inhibition reactions were proportional to the antithrombin–EP compound complex concentration in all cases, providing the second-order rate constants (kass) in Table 3. kass values were experimentally indistinguishable for reactions of any protease with antithrombin complexed with either EP42675 or EP217609, indicating no effect of the biotin tag. Comparison of kass values for all protease reactions showed that antithrombin–EP complexes preferentially inhibited factor Xa with a pharmacologically significant rate constant of ∼ 106M−1s−1 and with more than 20-fold selectivity over other proteases. This protease reaction profile resembled that of the antithrombin–idraparinux complex, except for proteases that were directly inhibited by the α-NAPAP-like part of the EP compound. The proteases that bound the α-NAPAP-like moiety with the highest affinity, thrombin, aPC, and factor XIa, showed either no detectable antithrombin–mediated inhibition by the EP compounds or a significantly reduced rate of inhibition relative to reactions with the antithrombin–idraparinux complex. This was fully accounted for by competitive inhibition of the antithrombin–mediated reaction by the α-NAPAP–like part of the EP compounds. Such competitive effects thus amplified the selectivity of the antithrombin–EP complex for inhibiting factor Xa relative to these proteases.
Stereoisomeric configuration of EP217609 is unimportant for direct and antithrombin-mediated inhibition of proteases
Because the biotin tag of EP217609 is attached at a chiral carbon, the compound consists of a mixture of 2 diastereoisomers, EP217609-1 and EP307138-1 (Figure 1). KI values and second-order rate constants for direct and antithrombin-mediated inhibition of proteases by the separate stereoisomers were therefore measured. The configuration of the biotin tag had minimal effect on the KD for binding of the stereoisomers to antithrombin (Table 1) or on KI values measured for direct inhibition of thrombin, aPC, plasmin, kallikrein, and factor Xa by the stereoisomers (Table 2). Only the direct inhibition of factor XIa was significantly affected by the biotin orientation, EP307138-1 binding with ∼ 4-fold higher affinity than EP217609-1 with the diastereomer mixture of EP217609 binding with intermediate affinity. Second-order rate constants for antithrombin-dependent inhibition of proteases by the EP stereoisomers similarly showed little or no significant differences between the stereoisomers for any of the proteases except for factor XIa (Table 3). The latter were accounted for by differential competitive inhibition by the direct inhibitor parts of the compounds.
Linkage effects on inhibitor potency
Because at plasma concentrations of antithrombin (2-3μM), EP217609 is expected to be fully complexed with the serpin, given the measured KD for this interaction, it was important to determine how antithrombin binding to the fondaparinux-like unit of the compound affects the binding of the α-NAPAP–like unit to proteases. This was assessed for the 3 proteases that bound the inhibitor with the highest affinity, namely, thrombin, aPC, and factor XIa. Comparison of titrations of these proteases with the EP compounds in the absence and presence of antithrombin levels that caused minimal antithrombin–mediated inhibition over the time of measurement showed that the serpin reduced the affinity of the EP compounds for the proteases from ∼ 2-fold for thrombin at 0.1μM antithrombin to 5- to 7-fold for aPC and factor XIa at 0.5 to 1μM serpin (Figure 5A).

Linkage effects on inhibitor potency of EP compounds. (A) Shown are the decrease in initial velocities of substrate hydrolysis for thrombin, aPC, and factor XIa measured as a function of increasing EP217609 concentration in the absence (solid symbols) or presence (open symbols) of 0.1, 0.5, and 1μM antithrombin, respectively. Solid and dashed lines indicate fits to the competitive binding equations in the text for titrations in the absence or presence of antithrombin, respectively. Values of KI* obtained from fits were corrected for competitive substrate binding for aPC and factor XIa. (B) Progressive inhibition of factor Xa by increasing concentrations of antithrombin–EP42675 (triangles) and antithrombin–EP217609 (circles) complexes was measured in the absence (open symbols) and presence (closed symbols) of thrombin levels equimolar with EP compound for reactions containing 200nM antithrombin, 0.5nM factor Xa, and the indicated EP concentrations for a fixed reaction time of 300 seconds. Inverted triangles indicate controls in which antithrombin was omitted from factor Xa reactions with equimolar EP42675 and thrombin. Solid lines indicate fits by a single exponential decay function, which yielded kobs values. These were used to calculate the dissociation constant for antithrombin binding to the EP-thrombin binary complex as described in “Linkage effects.”
Linkage effects on inhibitor potency of EP compounds. (A) Shown are the decrease in initial velocities of substrate hydrolysis for thrombin, aPC, and factor XIa measured as a function of increasing EP217609 concentration in the absence (solid symbols) or presence (open symbols) of 0.1, 0.5, and 1μM antithrombin, respectively. Solid and dashed lines indicate fits to the competitive binding equations in the text for titrations in the absence or presence of antithrombin, respectively. Values of KI* obtained from fits were corrected for competitive substrate binding for aPC and factor XIa. (B) Progressive inhibition of factor Xa by increasing concentrations of antithrombin–EP42675 (triangles) and antithrombin–EP217609 (circles) complexes was measured in the absence (open symbols) and presence (closed symbols) of thrombin levels equimolar with EP compound for reactions containing 200nM antithrombin, 0.5nM factor Xa, and the indicated EP concentrations for a fixed reaction time of 300 seconds. Inverted triangles indicate controls in which antithrombin was omitted from factor Xa reactions with equimolar EP42675 and thrombin. Solid lines indicate fits by a single exponential decay function, which yielded kobs values. These were used to calculate the dissociation constant for antithrombin binding to the EP-thrombin binary complex as described in “Linkage effects.”
To validate that the observed linkage of antithrombin binding and protease binding to the EP compounds was reciprocal, we evaluated the effect of thrombin at levels equimolar with EP42675 or EP217609 on the inhibition of factor Xa by the anti-thrombin–EP binary complex (Figure 5B). A modest, but highly reproducible, ∼ 30% decrease in the exponential rate constant for factor Xa inhibition was observed for both EP compounds in the presence of equimolar thrombin. Assuming that this decrease reflects a reduced affinity of the EP compound for antithrombin, we calculated an affinity loss of ∼ 5-fold (KD = 150nM; “Linkage effects”). This loss in antithrombin affinity suggested that the ∼ 2-fold decrease in EP affinity for thrombin observed at 100nM antithrombin underestimated the maximal affinity loss at saturating antithrombin. Calculation of the maximal affinity decrease from the linkage equation given in “Linkage effects” indicated an ∼ 5-fold loss in affinity of EP for thrombin at saturating levels of antithrombin, a value comparable to the loss in affinity observed with aPC and factor XIa at near-saturating antithrombin levels. The reciprocal ∼ 5-fold effects of thrombin binding on antithrombin binding and vice versa are consistent with the 2 interactions being thermodynamically linked. Notably, the linkage is modest, antithrombin binding to EP217609 still resulting in strong direct thrombin inhibition (KI ∼ 200pM) and thrombin binding minimally affecting the binding of antithrombin at plasma antithrombin concentrations (reduction from 99% to 93%).
Avidin effectively neutralizes the dual inhibitor moieties of EP217609
EP217609 contains a biotin tag that allows neutralization of the compound in vivo on avidin administration through accelerated clearance. To determine whether avidin binding directly affected the compound's dual inhibitor activities, we compared EP217609 inhibition of thrombin and factor Xa in the absence and presence of a 1- to 2-fold molar excess of avidin. Avidin markedly weakened the direct inhibitory effect of EP217609 on thrombin, the KI increasing 100-fold from 43 ± 16pM to 4.4 ± 0.3nM (Figure 6). Avidin had no effect on the KI for thrombin inhibition by the nonbiotinylated EP42675. The observation that the apparent KI measured in the presence of avidin was unaffected by doubling the substrate concentration indicated that no correction of this KI for substrate competition was necessary. This implied that avidin weakened the KI, not by increasing koff, but instead by decreasing kon.

Effect of avidin binding on EP217609 direct inhibition of thrombin. Titrations of the decrease in substrate hydrolysis activity (100μM S2366) of ∼ 1nM thrombin as a function of added EP42675 (circles) or EP217609 (triangles) in the absence (closed symbols) or presence (open symbols) of 50nM avidin. The effects of doubling the avidin or S2366 concentration in EP217609 titrations is shown by the open and closed inverted triangles, respectively. Titrations were fit by the equation for tight binding to obtain KI values.
Effect of avidin binding on EP217609 direct inhibition of thrombin. Titrations of the decrease in substrate hydrolysis activity (100μM S2366) of ∼ 1nM thrombin as a function of added EP42675 (circles) or EP217609 (triangles) in the absence (closed symbols) or presence (open symbols) of 50nM avidin. The effects of doubling the avidin or S2366 concentration in EP217609 titrations is shown by the open and closed inverted triangles, respectively. Titrations were fit by the equation for tight binding to obtain KI values.
Avidin reduced kobs for EP217609-antithrombin complex inhibition of factor Xa by ∼ 5-fold but had no effect on EP42675-antithrombin complex inhibition (Figure 7A). To verify that this was the result of a reduced affinity of antithrombin for the EP217609-avidin complex, we performed kinetic titrations of antithrombin with free and avidin-complexed EP217609 under conditions where saturable increases in kobs for antithrombin inhibition of factor Xa provided the KD for EP compound binding to antithrombin as well as the maximal rate constant associated with full activation of the serpin (Figure 7B).34 Avidin substantially weakened the binding of EP217609 to antithrombin without affecting the maximal activation. The KD values obtained in the absence and presence of avidin equimolar with the highest EP compound concentration assuming equivalent endpoints were 44 ± 6nM and 1040 ± 50nM, respectively, indicating that avidin produces a 20- to 30-fold reduction in EP217609 affinity for antithrombin. The observed ∼ 5-fold reduction in the apparent EP-accelerated rate constant but ∼ 20- to 30-fold reduction in EP affinity for antithrombin is a consequence of the antithrombin concentration used to measure the rate constant exceeding the KD for EP compound binding in the absence of avidin but being well below KD in the presence of avidin. When the apparent rate constants measured are corrected for the increase in KD caused by avidin, equivalent second-order rate constants of 7.6 ± 1.2× 105M−1s−1 and 7.7 ± 0.9 × 105M−1s−1 for the antithrombin–EP217609 complex reaction with factor Xa in the absence and presence of avidin, respectively, were obtained.

Effect of avidin on EP217609-accelerated antithrombin inhibition of factor Xa. (A) EP217609-accelerated antithrombin inhibition of factor Xa activity is shown as a function of EP217609 concentration in the absence and presence of avidin. Reactions contained 200nM antithrombin, 5nM factor Xa, the indicated EP compound concentration, and 0 (●), 12nM (▴), or 24nM (▵) avidin as indicated for a fixed reaction time of 300 seconds. Solid lines indicate fits by a single exponential function from which kobs was obtained. (B) Kinetic titrations of the binding of EP217609 to 50nM antithrombin in the absence (●) and presence (▴) of 1000nM avidin followed from the enhancement by the EP compound of the rate of antithrombin inhibition of 5nM factor Xa. Plotted are observed pseudo-first-order rate constants (kobs) calculated from the extent of antithrombin inhibition of factor Xa in the presence of the indicated concentrations of EP217609 in the absence or presence of avidin for 1-minute reactions. Solid lines indicate fits to the quadratic binding equation from which KD for the antithrombin–EP217609 interaction and the maximal rate constant for the antithrombin–EP217609 reaction with factor Xa were obtained. It should be noted that reciprocal effects of antithrombin binding on avidin binding are insignificant because avidin binds biotin with a femtomolar KD.
Effect of avidin on EP217609-accelerated antithrombin inhibition of factor Xa. (A) EP217609-accelerated antithrombin inhibition of factor Xa activity is shown as a function of EP217609 concentration in the absence and presence of avidin. Reactions contained 200nM antithrombin, 5nM factor Xa, the indicated EP compound concentration, and 0 (●), 12nM (▴), or 24nM (▵) avidin as indicated for a fixed reaction time of 300 seconds. Solid lines indicate fits by a single exponential function from which kobs was obtained. (B) Kinetic titrations of the binding of EP217609 to 50nM antithrombin in the absence (●) and presence (▴) of 1000nM avidin followed from the enhancement by the EP compound of the rate of antithrombin inhibition of 5nM factor Xa. Plotted are observed pseudo-first-order rate constants (kobs) calculated from the extent of antithrombin inhibition of factor Xa in the presence of the indicated concentrations of EP217609 in the absence or presence of avidin for 1-minute reactions. Solid lines indicate fits to the quadratic binding equation from which KD for the antithrombin–EP217609 interaction and the maximal rate constant for the antithrombin–EP217609 reaction with factor Xa were obtained. It should be noted that reciprocal effects of antithrombin binding on avidin binding are insignificant because avidin binds biotin with a femtomolar KD.
Discussion
Previous reports have documented the potent anti-IIa and anti–factor Xa activities of EP217609 and its nonbiotinylated counterpart EP42675 in plasma, as well as the favorable pharmacokinetic properties, sustained antithrombotic activity, and avidin-mediated neutralization of EP217609 in vivo.15,17 In the present study, we have performed a comprehensive biochemical characterization of EP217609 and EP42675 potency and selectivity to provide new mechanistic insights into the potent antithrombotic activity of EP217609. Our results show that the inhibitors possess a high degree of specificity for their targets (KI = 30-60pM for thrombin and kass 6 to 7 × 105M−1s−1 for factor Xa) as well as a marked selectivity for these proteases (KI > 1000-fold greater and kass > 20-fold slower for all other coagulation and fibrinolytic proteases tested). The biotin tag was found to minimally perturb the specificity and selectivity toward the dual targets in either of its stereoisomeric configurations, thereby providing a nonperturbing means for neutralizing the compound's activity with avidin. Notably, avidin binding to EP217609 was found to cause a large loss in inhibitor potency (100-fold increase in KI for thrombin and 20- to 30-fold increase in the KD for antithrombin binding), contributing to an immediate neutralization of the drug's activity in addition to the indirect neutralization caused by avidin accelerating its clearance.15,35 Phase 1 clinical studies in humans have indeed shown that administration of avidin triggers a rapid and irreversible neutralization of EP217609 without rebound effect.18
The selectivity of EP217609 as a direct thrombin inhibitor largely derives from the extremely high affinity of the α-NAPAP–like moiety for thrombin (KI = 30-40pM), which ranks among the highest-affinity direct thrombin inhibitors known.6,36 This affinity is much greater than that of α-NAPAP or related inhibitors, such as argatroban,37 the result of structure-activity optimization that included replacement of the naphthylsulfonamide moiety and incorporation of the active stereoisomer of the amidinophenyl substituent.14 The kinetics of inhibition are rapid and conform to a simple one-step reversible equilibrium with no evidence for any induced-fit slow binding step. The protease with the next highest affinity, aPC, was inhibited with a KI value of ∼ 10−7M, which was 1000-fold higher than that for thrombin. Factor XIa was inhibited with an even higher KI approaching micromolar, and all other proteases were inhibited with KI values exceeding 10−6M. This selectivity parallels that previously reported for other α-NAPAP analogs, although those with considerably reduced specificity for thrombin.36 The biotin tag somewhat enhanced the affinity for all proteases except aPC, thereby contributing to the high selectivity of EP217609 for thrombin over aPC. Because EP217609 is expected to act pharmacologically with antithrombin bound, it was important to find that antithrombin binding only modestly decreased the affinity of EP217609 for thrombin without affecting the selectivity for thrombin over aPC. Therapeutic doses of EP217609 are thus not expected to significantly perturb the anticoagulant function of aPC.
EP217609 and the nonbiotinylated EP42675 were found to act like the natural heparin pentasaccharide fondaparinux or its higher-affinity mimetic idraparinux in transforming antithrombin into a highly selective factor Xa inhibitor.25 Thus, comparison of the relative values of kass for reaction of proteases with antithrombin complexed with any of the EP compounds showed that EP compound activation of antithrombin, as with fondaparinux or idraparinux activation, alters the specificity profile of antithrombin to favor inhibition of factor Xa over all other proteases. Factor Xa is thus inhibited by the antithrombin–EP compound complex with a rapid rate constant of ∼ 106M−1s−1, whereas all other coagulation and fibrinolytic proteases whose inhibition rate could be measured are inhibited 20- to 50 000-fold slower than factor Xa.
Importantly, the 2 inhibitor moieties of EP217609 were found to act in a largely independent manner with relatively modest ∼ 5-fold linkage effects of the binding of one protein ligand to the EP compound on the binding of the other. Either inhibitor moiety thus retains a high potency irrespective of whether the other moiety is bound to its protein ligand. The flexible spacer unit that separates the 2 inhibitor moieties thus allows each inhibitor to act independently. By contrast, placement of the biotin tag at the junction between the 2 inhibitor moieties results in a larger effect of avidin binding on the inhibitor interactions with their protein ligands.
The present report together with prior reports shows that the strategy of combining 2 separate inhibitor moieties with distinct mechanisms of action yields an anticoagulant with unprecedented pharmacologic properties. Thus, the predictable pharmacokinetic properties of the fondaparinux analog are transferred to a direct thrombin inhibitor, making EP217609 a unique anticoagulant. Its potent antithrombotic activity in both venous and arterial thrombosis models is in part related to its ability, shared with other direct thrombin inhibitors, to inactivate clot-bound thrombin as well as free thrombin. Importantly, its long half-life, predictable pharmacokinetics, and neutralizability cannot be replicated by coadministration of the individual inhibitor compounds. EP217609 thus shows great promise in overcoming the limitations of current anticoagulants, and the EP217609-avidin couple represents an attractive anticoagulation system for CPB. Ongoing clinical trials will determine whether this new system offers a better benefit/risk ratio than the heparin-protamine system that has been used for more than 60 years in this indication.
There is an Inside Blood commentary on this article in this issue.
Acknowledgments
The authors thank Dr Peter Gettins of the University of Illinois at Chicago for helpful comments on the manuscript.
This work was supported in part by a contract with Endotis Pharma and by the National Institutes of Health (grant R37 HL-39888; S.T.O.).
National Institutes of Health
Authorship
Contribution: S.T.O. and M.P. designed the research study; R.S. acquired the research data; R.S. and S.T.O. analyzed and interpreted data; and S.T.O. and M.P. wrote the manuscript.
Conflict-of-interest disclosure: M.P. is employed by Endotis Pharma as Head of Preclinical Research and Development. The remaining authors declare no competing financial interests.
Correspondence: Steven T. Olson, Center for Molecular Biology of Oral Diseases, University of Illinois at Chicago, 801 S Paulina St, Rm 530C, Chicago, IL 60612; e-mail: stolson@uic.edu.

