Abstract
Cardiovascular diseases, including atherothrombosis, are the leading cause of morbidity and mortality in the United States, Europe, and the developed world. Matrix metalloproteases (MMPs) have recently emerged as important mediators of platelet and endothelial function, and atherothrombotic disease. Protease-activated receptor-1 (PAR1) is a G protein-coupled receptor that is classically activated through cleavage of the N-terminal exodomain by the serine protease thrombin. Most recently, 2 MMPs have been discovered to have agonist activity for PAR1. Unexpectedly, MMP-1 and MMP-13 cleave the N-terminal exodomain of PAR1 at noncanonical sites, which result in distinct tethered ligands that activate G-protein signaling pathways. PAR1 exhibits metalloprotease-specific signaling patterns, known as biased agonism, that produce distinct functional outputs by the cell. Here we contrast the mechanisms of canonical (thrombin) and noncanonical (MMP) PAR1 activation, the contribution of MMP-PAR1 signaling to diseases of the vasculature, and the therapeutic potential of inhibiting MMP-PAR1 signaling with MMP inhibitors, including atherothrombotic disease, in-stent restenosis, heart failure, and sepsis.
Introduction
Protease-activated receptors (PARs) are members of the G-protein coupled receptor superfamily whose cellular responses are driven through various G-protein and non–G-protein pathways, resulting in a diverse array of physiologic outputs.1 PARs were identified > 20 years ago with the discovery of the first thrombin receptor,2,3 later renamed PAR1. Elucidation of the unusual proteolytic mechanism of receptor activation paved the way for the subsequent discovery of PAR2, PAR3, and PAR4.4 After the observation that human platelets could be activated by proteolytic cleavage of PAR1 by thrombin, many other members of the serine protease family were found to be capable of activating one or another of the PARs, including plasmin, activated protein C, thrombocytin, PA-BJ, factor Xa, factor VIIa, kallikreins, cathepsin G, trypsin, matriptase, and tryptase.5-8 Considering that PARs were initially identified as thrombin receptors, their role in the vasculature is paramount.1 PARs are expressed on nearly all cell types in the blood vessel wall and blood, the notable exception being red blood cells.1 PAR1 is the high-affinity thrombin receptor and is expressed on the surface of endothelium, smooth muscle cells, platelets, neutrophils, macrophages, and leukemic white cells.9,10 Thrombin activation of PAR1 promotes platelet aggregation, shape change, adhesion, cell proliferation, chemokine production, and migration via Gq, Gi, and G12/13 pathways.11
In this review, we focus on the unexpected discovery that the zinc-dependent matrix metalloprotease-1 (MMP-1) is able to cleave and activate PAR1 at a noncanonical site,12 which leads to a signaling pattern in platelets and other cells distinct from that seen with thrombin. Platelets harbor abundant proMMP-1 zymogen on their surface,13 which is converted to active MMP-1 after exposure to collagen fibrils.12 Recently, a second MMP, MMP-13, was identified as having the capacity to cleave and activate PAR1 on cardiac fibroblasts and cardiomyocytes, resulting in pathologic activation of downstream signaling events that contribute to heart failure.14 Emerging evidence suggests that selective proteolytic activation of PAR1 by MMPs, such as MMP-1 and MMP-13, will be important contributors to the evolution of a variety of disease processes, including thrombus initiation and thrombosis, atherosclerosis and restenosis, sepsis, angiogenesis, heart failure, and cancer (Figure 1).

MMP-PAR1 signaling in vascular diseases. PAR1 senses a diverse milieu of extracellular proteases and subsequently relays that information to influence cellular behavior and potentially exacerbate disease pathologies. The N-terminal extracellular domain (exodomain) of PAR1 is cleaved at a canonical site by thrombin and noncanonical sites by MMP-1 and MMP-13. Various signaling outputs can lead to platelet thrombosis, atherosclerosis, in-stent restenosis, heart failure, and sepsis.
MMP-PAR1 signaling in vascular diseases. PAR1 senses a diverse milieu of extracellular proteases and subsequently relays that information to influence cellular behavior and potentially exacerbate disease pathologies. The N-terminal extracellular domain (exodomain) of PAR1 is cleaved at a canonical site by thrombin and noncanonical sites by MMP-1 and MMP-13. Various signaling outputs can lead to platelet thrombosis, atherosclerosis, in-stent restenosis, heart failure, and sepsis.
Divergence in the MMP family
MMPs compose a family of 28 zinc-dependent endopeptidases, which are further subdivided based on their distinct, albeit overlapping, substrate specificity.15 MMP-1, -8, and -13, otherwise known as the interstitial collagenases, are capable of initiating the degradation of fibrillar-type collagens by cleaving at a single site three-fourths of the way from the N-terminus.16 Because collagen is the most abundant protein in the human body, the collagenases represent an essential enzyme class involved in normal development and tissue repair.17 MMP-2 and -9 compose the gelatinases, the primary enzymes capable of degrading gelatinous byproducts of collagen degradation.18 MMP-3, -7, -10, and -11 are members of the stromelysin subfamily, which degrade laminin, fibronectin, and elastin, among others.19 MMP-12 is thought to be a distantly related metalloelastase, predominantly found in macrophages, which primarily degrades elastin.18,20 The final class is composed of the membrane-tethered MMPs; these include MMP-14, -15, -16, -17, -24, and -25.21
Nearly all MMPs are secreted as zymogens and acquire activity through removal of their autoinhibitory prodomain (Figure 2A).22 The prodomain of MMPs contains a highly conserved cysteine residue responsible for coordinating with the active-site zinc to lock the enzyme in an inactive state. Disruption of the cysteine-zinc interaction, either through chemical modification or by proteolytic release of the prodomain, results in activation of the zymogen.22

MMP-PAR1 interactions. (A) Structure of the proMMP1 zymogen. The catalytic zinc is indicated as a red ball within the catalytic domain. (B) Cleavage sites for MMP-1, MMP-13, and thrombin in PAR1 and resulting tethered ligands. (C) Top: N-terminal region encompassing the PAR1-tethered ligand showing the cleavage sites for MMP-1 and thrombin. Bottom: Structural model of the catalytic groove of MMP-1 bound to the N-terminal domain of PAR1, illustrating the catalytic zinc coordinated to the PAR1 scissile D-P bond. The model was created using the x-ray structures of MMP-1 and MMP-8 bound to peptide inhibitors as templates.49,50
MMP-PAR1 interactions. (A) Structure of the proMMP1 zymogen. The catalytic zinc is indicated as a red ball within the catalytic domain. (B) Cleavage sites for MMP-1, MMP-13, and thrombin in PAR1 and resulting tethered ligands. (C) Top: N-terminal region encompassing the PAR1-tethered ligand showing the cleavage sites for MMP-1 and thrombin. Bottom: Structural model of the catalytic groove of MMP-1 bound to the N-terminal domain of PAR1, illustrating the catalytic zinc coordinated to the PAR1 scissile D-P bond. The model was created using the x-ray structures of MMP-1 and MMP-8 bound to peptide inhibitors as templates.49,50
To date, only 3 MMPs have been identified as having agonist activity against PAR1: MMP-1, Mmp-1a, and MMP-13.14,23-25 MMP-1, Mmp-1a, and MMP-13 all belong to the collagenase subfamily and share several important structural features, including classic signal peptide, pro-, catalytic, and hemopexin domains characteristic of this subgroup (Figure 2A). MMP-1 is expressed in most human tissues, including the majority of cell types in the blood vessel wall, inflammatory cells, and platelets, and is considered the primary enzyme responsible for collagen degradation.13,26,27 Despite the pervasive basal expression of MMP-1, a number of disease states result in further up-regulation of MMP-1, a consequence that is often associated with poor outcomes.18,27,28 Mmp-1a, the mouse ortholog of human MMP-1, is expressed in similar cell types as human MMP-1, albeit to a lesser extent, but is also up-regulated in various diseases, including inflammation, sepsis, and cancer.24,25,29
MMP-13 is more limited in its tissue expression profile compared with MMP-1 and is important to developing bone and periodontal tissues.30,31 Similar to MMP-1, MMP-13 is up-regulated in a number of pathologic states, including atherosclerosis, rheumatoid arthritis, periodontitis, breast cancer, melanoma, and squamous cell carcinomas of the head and neck.27,32
Although MMPs are classically categorized based on their extracellular-matrix substrate specificity, they also function as important signaling molecules through the cleavage of > 100 extracellular ligands and proteins.15 Important nonmatrix substrates include osteopontin,33 SDF-1α,34 HB-EGF,35 IL-1β,36 and now PAR1.
Canonical thrombin-PAR cleavage
Cleavage of the extracellular portion of the PAR1 receptor by thrombin occurs at a canonical R41-S42 site, which is distinct from the MMP-1 and MMP-13 cleavage sites (Figures 1 and 2C). The freshly cleaved N-terminus is then capable of interacting with the C-terminal portion the exodomain37 and other extracellular loops,38 resulting in conformational changes in the transmembrane domain/intracellular loops and subsequent signal transduction.39 Exogenous addition of synthetic peptides corresponding to the thrombin-generated tethered ligand (eg, SFLLRN, Figure 2B) are able to stimulate signaling outputs that mimic proteolytic activation of the receptor,2 thus providing critical evidence that PARs carry their own ligands.
Thrombin cleavage of PAR1 is facilitated by a K51YEPF55 motif that resembles the C-terminus of the leech anticoagulant protein hirudin.37 Binding of exosite I of thrombin to the hirudin-like sequence of PAR1 results in allosteric changes to thrombin, which lowers the activation energy necessary for cleavage at the LDPR-S canonical site on PAR1.40 Mutational analysis of the thrombin cleavage site identified the P4-Leu38 and P2-Pro40 residues as critical for proper cleavage of PAR1.41,42 By comparison, the low-affinity PAR4 receptor lacks the functional hirudin-like sequence found in PAR1 and does not bind exosite I to cause allosteric activation of thrombin.43 Instead, PAR4 contains an anionic cluster, Asp57 Asp59 Glu62 Asp65 in its exodomain, which slows the dissociation rate of PAR4 from the cationic thrombin.43 Although PAR4 is cleaved more slowly than PAR1,44 polypeptides containing the PAR4 P2-P1 active site-interacting sequence, Pro45-Ala-Pro-Arg (PAPR), are efficiently cleaved because of the optimal placement of dual prolines at positions P4 and P2.43
Noncanonical MMP-PAR1 cleavage and biased agonism
Recently, it was found that PAR1 is also substrate for the collagenases, MMP-1 and MMP-13 (Figure 2), which cleave PAR1 at noncanonical sites distinct from thrombin.12,14 The initial identification of the MMP-1 cleavage site of PAR1 stemmed from cleavage studies using a synthetic 26 amino-acid peptide (TR26), which spanned the N-terminal tethered ligand region of PAR1.12 Incubation of nanomolar MMP-1 with TR26 yielded a peptide with a mass that corresponded to cleavage at the D39-P40 peptide bond. This cleavage resulted in a tethered ligand, which is 2 amino acids longer than the thrombin-generated ligand. To confirm that this cleavage event would result in productive PAR1 signaling, the longer MMP-1 ligand PR-SFLLRN peptide was synthesized and tested for platelet activity. The result was complete recapitulation of the signaling cascades activated by the authentic MMP-1 enzyme, including platelet shape change, RhoA activation, and p38 phosphorylation, but was a relatively weak agonist for stimulating intracellular calcium mobilization and platelet aggregation. These effects were all blocked by a small-molecule inhibitor of PAR1, RWJ-56110, confirming that this was a PAR1-dependent event.12 Together, these data demonstrated that MMP1-PAR1 rapidly activated Rho-GTP pathways, cell shape change, motility, and MAPK signaling in platelets.12 Subsequent analysis of G protein signaling pathways presented the first evidence that MMP-1 and the PR-SFLLRN ligand are biased agonists that preferentially activate G12/13, and thrombin preferentially activates Gq in human platelets.12
Blackburn and Brinckerhoff45 showed that MMP-1 and thrombin differentially activate MAPK signaling in endothelial cells, with MMP1-driven signals delayed (15 minutes) compared with thrombin (5 minutes), both of which were inhibited by a PAR1 specific antagonist. In breast cancer cells, thrombin and MMP-1 activated PAR1-dependent phospho-Akt signals,46 with the peak Akt signal occurring at 5 minutes for thrombin and 1 hour for MMP-1, showing that, even with identical signaling outputs, thrombin and MMP1 can display divergent kinetics.46 Perhaps more striking was the differential effects seen on PAR1-dependent gene expression of certain proangiogenic factors in endothelial cells.45 The 2 enzymes individually increased expression of subsets that were independent of the other protease, suggesting that MMP1-PAR1 signaling may alter the phenotype of cells in a distinct manner from thrombin-PAR1 activation.45
Although thrombin is specific for the R41↓S42FLLRN bond, MMP-1 generally prefers a hydrophobic residue at the C-terminal side (P1′) of the cleavage site, a basic or hydrophobic amino acid at P2′, and a small residue (alanine, glycine, or serine) at P3′, which facilitates proper fit between the substrate and the S1′-S3′ sites on the enzyme surface.47,48 By placing the hydrophobic P40 residue at the P1′ position, the alignment of the PAR1 cleavage peptide resembles that seen in native substrates of the collagenases.47,48 Using x-ray structures of MMP-1 and MMP-8 bound to peptide inhbitors,49,50 we modeled the PAR1 ligand region consisting of the residues T37LD-PRSFLLRN47 bound to the active site cleft of human MMP-1 with the scissile D39-P40 peptide bond carbonyl oxygen coordinated to the active site zinc (Figure 2C). Especially favorable hydrophobic interactions and geometry were observed for PAR1 P3 and P2 residues (T37, L38) and for the C-terminal cleavage site P1′-P40 within the active site groove surrounding the catalytic zinc of MMP-1. To provide evidence to support this model, the P1′-P40 residue was substituted with asparagine in both the full-length receptor (P40N PAR1) and the cleavage peptide (TR26-P40N), a substitution that had previously been shown to substantially reduce cleavage of collagen peptides.51 To inhibit proteolysis by thrombin, the P1′-S42 serine of the thrombin cleavage site was mutated to aspartate (S42D PAR1), a mutation that suppresses cleavage by thrombin.52 The P40N PAR1 mutant was fully cleaved by thrombin but was poorly cleaved by MMP-1.12 Conversely, the S42D PAR1 mutant was substantially cleaved by MMP-1 but was poorly cleaved by thrombin. These results were consistent with the mass spectrometry cleavage data that MMP-1 cleaves PAR1 at D39-P40 and illustrated the ability to selectively uncouple MMP-1 cleavage of PAR1 from thrombin.12
Two other MMP-1 cleavage sites in the PAR1 N-terminal exodomain have been described. Nesi and Fragai53 showed that, using a longer incubation time and MMP-1 at micromolar concentrations, recombinant PAR1 (A26-L103) exodomain5 was cleaved at the L44-L45 and F87-I88 bonds, which could result in desensitized receptor because of loss of part or all of the tethered ligand. As the L44-L45 and F87-I88 MMP1-cleaved receptors have not been validated physiologically, it is unclear whether MMP-1 cleavage sites within or distal to the canonical ligand are capable of productive signaling. However, it was recently shown that activated protein C may also cleave PAR1 at R46-N47,54 as opposed to the canonical R41-S42 cleavage site,5 similar to the snake venom proteases thrombocytin and PA-BJ.6 Although cleavage at the C-terminus of the tethered ligand might be expected to severely impact signaling, the activated protein C R46-N47 cleavage event is capable of producing cytoprotective effects through PAR1, representing a new example of biased agonism for PAR1.54 This raises the question of whether additional proteases that cleave at distal sites may produce new kinds of signaling outputs from PARs either as homomers or in partnership with other PARs in heteromeric complexes, such as PAR1-PAR2 heterodimers.55,56 It is also possible that conformational changes or steric steering, which might occur through dimerization, could promote one cleavage site over another. Once cleaved, the different-length tethered ligands may induce or stabilize different conformational states in the transmembrane domains and intracellular loops of PAR1 to facilitate preferential or biased association with one class of G-proteins over another.57,58 Moreover, enzyme coreceptor proteins, including integrins, or local microenvironments and lipid rafts,59 could favor biased cleavage and subsequent activation of precoupled58 subsets of receptors.
Most recently, MMP-13 was identified as having PAR1 agonist activity.14 Using similar cleavage experiments performed before with MMP-1, the TR26 PAR1 peptide was found to be cleaved by MMP-13 at a slightly different site, S42-F43, one residue toward the C-terminal side of the thrombin cleavage site. This cleavage site was functionally validated through mutation of amino acid F43, rendering the receptor insensitive to MMP-13 cleavage.14 Considering that the MMP family has a conserved active site,20 it is possible that other MMPs are also capable of cleaving PAR1. Several other MMPs that were tested for PAR1 cleavage and/or signaling, including MMP-2, -3, -7, and -9, appeared to be unable to activate PAR1,12,23 although many other MMPs have yet to be tested for agonist activity against PAR1 or the other PARs.
MMP1-PAR1 in platelet activation and atherothrombosis
Platelets express several metalloproteases, including MMP-1, MMP-2, MMP-3, and MMP-14, on their surface13,60,61 (Figure 3), and megakaryocytes selectively transfer specific MMP mRNAs and proteins into platelets.62 Pioneering studies from nearly 40 years ago had shown that platelets harbored collagenases capable of degrading fibrillar collagen.63 The platelet collagenase activity was later identified as MMP-1, which was released after exposure of platelets to thrombin.13 More recently, it was discovered that collagen-stimulated platelet activation also resulted in the conversion of proMMP-1 to active MMP-1, which was subsequently capable of directly cleaving PAR1 on the platelet surface.12 Coimmunoprecipitation experiments indicated that proMMP-1 forms a stable complex with the α2β1 and αIIbβ3 integrins on platelets. MMP1-PAR1 signaling in platelets led to activation of G12/13-Rho, p38 MAPK pathways, and platelet shape change. Furthermore, the collagen-MMP1-PAR1 pathway was able to mediate platelet thrombogenesis and clot retraction, which was inhibited by PAR1 antagonists, but not thrombin inhibitors.12 These results suggested that the collagen-MMP1-PAR1 pathway is an activator of both early and late platelet signaling events independent of thrombin.

MMP and PAR1 expression in atherothrombotic disease. Specific MMPs and PAR1 and their cellular sources in an atherosclerotic blood vessel. Instability of the fibrous cap overlaying the plaque can lead to subendothelial collagen exposure and subsequent platelet activation. Platelets express MMP-1, -2, -3, and -1413,60,61 and exposure of naive platelets to collagen activates MMP-1 from the proMMP1 zymogen. Endothelial cells express a number of MMPs, including MMP-1, -2, -9, -13, and -14.18 Macrophages and smooth muscle cells (SMCs) are activated (Mφ foam cells and SMCs with red nuclei) by the proinflammatory state resulting from cholesterol deposition in the plaque body and secrete MMP-1, -2,-9, and -13, among others.18,26
MMP and PAR1 expression in atherothrombotic disease. Specific MMPs and PAR1 and their cellular sources in an atherosclerotic blood vessel. Instability of the fibrous cap overlaying the plaque can lead to subendothelial collagen exposure and subsequent platelet activation. Platelets express MMP-1, -2, -3, and -1413,60,61 and exposure of naive platelets to collagen activates MMP-1 from the proMMP1 zymogen. Endothelial cells express a number of MMPs, including MMP-1, -2, -9, -13, and -14.18 Macrophages and smooth muscle cells (SMCs) are activated (Mφ foam cells and SMCs with red nuclei) by the proinflammatory state resulting from cholesterol deposition in the plaque body and secrete MMP-1, -2,-9, and -13, among others.18,26
In addition to the role of MMP-1 in platelet activation, the matrix-degrading functions of MMP-1 in the vessel wall are critical.32 In particular, there has been an increasing awareness of the diagnostic value of plasma MMP-1 in patients with acute coronary syndromes (ACS) and its surge after percutaneous coronary intervention (PCI) and stenting.64 Plasma MMP-1 is also significantly increased in diabetic patients and those with high intimal/medial ratios in their carotid artery plaques.65 Although pathoanatomic studies of human atherosclerotic lesions suggest that large plaques cause ischemic symptoms, the key contributing factor to the morbidity and mortality associated with atherosclerosis is excessive platelet thrombus formation on exposed collagen surfaces after acute plaque rupture.66 The contribution of MMP-1 during atherogenesis can thus be considered 2-fold: destabilization of the collagenous structure of plaques in addition to a procoagulant function provided by its effect on platelets.
MMP-1 expression is increased in atherosclerotic plaques.26,32,67 Furthermore, MMP-1 has been shown to be to be expressed by macrophages, smooth muscle cells, and endothelial cells surrounding the fibrous cap, especially in the vulnerable shoulder region of the plaque.67,68 In addition, PAR1 expression is increased in the plaque body,69 suggesting that the activating enzyme (MMP-1) and receptor (PAR1) would be in close proximity during plaque remodeling. Although the direct relationship between MMP-1 and PAR1 in the atherosclerotic plaque has not yet been shown, genetic deletion of PAR1 or inhibition regulates neointimal hyperplasia after vessel damage in animal models.70-72 Furthermore, as tissue factor may also be increased in atherosclerotic lesions, the contributions of thrombin-PAR1 versus MMP-PAR1 signaling in this context have not been examined. Future studies should be aimed at determining the functional agonist in these settings and whether direct inhibition of the protease would have any beneficial effect.
Similar to MMP-1, MMP-13 is also up-regulated in atherosclerotic plaques.32 A study by Deguchi et al, using MMP-13 knockout mice bred onto an ApoE−/− background, showed no significant differences in plaque formation or composition.73 On the contrary, a study performed in mice lacking the LDL-receptor showed an 11-fold increase in MMP-13 expression in the aortas of atherosclerotic animals.74 Additional studies will be useful to help clarify the role of MMP-13 in atherosclerotic plaque formation and resolution.
MMP13-PAR1 in heart failure
PAR1 may also play an important role in cardiovascular development and cardiac hypertrophy.75-78 PAR1 (f2R) knockdown results in a weaker heartbeat and slower blood flow in zebrafish.78 Conversely, PAR1 stimulation results in cardiomyocyte hypertrophy and increased DNA synthesis in cardiac fibroblasts.76 Cardiac-specific overexpression of PAR1 led to heart failure in mice, whereas genetic deletion of PAR1 blunted the injury response after ischemia and reperfusion.77 It was recently suggested that the proteolytic agonist for the observed PAR1-dependent cardiac phenotype in mice was MMP-13.14 MMP-13 is expressed in normal adult heart tissues and is increased in cardiac fibroblasts after β-adrenergic receptor activation, a common sequelae in early heart failure. The newly released MMP-13 was able to cleave and activate PAR1 on neonatal rat ventricular myocytes.14 As PAR1 was cleaved by MMP-13 at a distinct site from both MMP-1 and thrombin, at S42-F43, it will be interesting to determine whether this leads to specific signaling pathways that exacerbate cardiac dysfunction. Finally, it was shown that either genetic deletion of PAR1 or inhibition of MMP-13 could prevent the deleterious cardiac effects of β-adrenergic receptor overstimulation.14 These results indicate that sustained activation of MMP13-PAR1 in cardiac tissue may be a maladaptive response in heart failure models.
MMP1-PAR1 in sepsis
Sepsis and septic shock represent a cohort of disease states in which dysregulation of the vasculature is a key pathologic feature. The coincident activation of proinflammatory mediators contributes to endothelial barrier disruption and subsequent hypotension, hypovolemia, and disseminated intravascular coagulation (DIC).79,80 Endothelial PAR1 may also be a major mediator of acute inflammatory-coagulation responses and is a potential target in disease states characterized by endothelial dysfunction, including abnormal hypercoagulability and sepsis/systemic inflammatory response syndrome.55,81,82 Thrombin activation of PAR1 causes Rho-dependent cytoskeletal rearrangements in endothelial cells and induces cell contraction and rounding.83,84 Endothelial cell contraction destabilizes cell-cell contacts causing a subsequent increase in vascular permeability, which facilitates the passage of molecules and leukocytes from the blood into subendothelial compartment and exposure of tissue factor and collagen, which can trigger DIC. Despite the ability of thrombin to trigger PAR1-dependent activation of endothelial cell contraction, PAR1 signaling confers both beneficial and deleterious effects on sepsis progression and outcomes depending on the timing and the severity of the disease state.55,85
Recently, it was shown that MMP1-PAR1 signaling also plays an important role in endothelial barrier function and sepsis outcomes.24 Sepsis patients had a significant 18-fold increase in mean levels of proMMP-1 in their plasma at time of enrollment relative to healthy controls. Elevated proMMP-1 levels significantly correlated with worsening survival outcomes at day 7 and 28 for septic patients (P = .006).24 In mouse models of sepsis, mouse Mmp-1a was released from the endothelium into the circulation. Both Mmp-1a and MMP-1 triggered PAR1-dependent disruption of barrier function via Rho pathways. Inhibition of MMP-1 in the early stages of sepsis significantly improved the survival of WT and PAR2−/− mice but had no effect in PAR1−/− mice.24 Similar results have been observed with thrombin inhibition in endotoxemia models using WT and PAR2−/− mice.85 Administration of exogenous human MMP-1 caused endothelial barrier dysfunction and increased lung vascular permeability in WT but not PAR1−/− mice.24 Conversely, sepsis- and lipopolysaccharide-induced vascular leakage could be attenuated by inhibition of MMP-1 activity. Inhibition of MMP-1 also reduced DIC and markedly suppressed the cytokine storm, which was lost in PAR1−/− mice. The timing of the effects of inhibition of MMP-1 activity on lung vascular permeability, systemic cytokines, and DIC correlated well with survival outcomes in mice.24 These findings suggest that endothelial MMP1-PAR1 plays an unexpectedly important role in the lethal sequelae of sepsis and that MMP-1 could be a useful predictive biomarker for outcomes in patients newly diagnosed with sepsis.
Therapeutic potential of inhibiting MMPs in thrombosis and cardiovascular disease
Activation of platelet thrombosis in patients with acute coronary syndromes often occurs under high shear-stress conditions on subendothelial surfaces enriched in collagen fibrils.66 Studies using human whole blood spiked with either MMP-1 or PAR1 inhibitors, such as a PAR1 pepducin,86 did not affect primary adhesion of platelets to immobilized collagen fibrils under arterial shear.12 However, the growth rate of platelet aggregate “strings” was significantly attenuated by an MMP-1 inhibitor, FN-439, or PAR1 inhibitors. Compared with MMP-1 inhibition, antagonism of thrombin had little effect on early thrombogenesis on collagen surfaces under high arterial flow rates.12 Several studies have previously shown that thrombin may be more important for later propagation and stability of platelet thrombi and is not involved in initiating early thrombus growth at high arterial shear,87-89 unless tissue factor levels are extremely high.90 Blockade of the MMP1-PAR1 pathway with the MMP-1 inhibitor, FN-439, also greatly curtailed arterial thrombosis in a guinea pig model of ferric chloride injury (Table 1), which causes denudation of the artery and exposure of type I collagen and other subendothelial matrix proteins.12 These in vitro and in vivo data suggest that the collagen-MMP1-PAR1 pathway may be a point of early intervention in preventing arterial thrombosis.
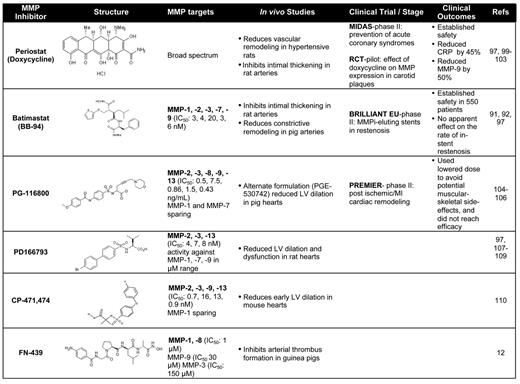
Considering the complex role of MMPs in vascular remodeling, especially in intimal thickening after balloon angioplasty and stenting,91,92 the first MMP inhibitor (MMPi) clinical trials in the cardiovascular field were used to target restenosis (Table 1). Additional evidence from animal models suggested that genetic deletion of MMPs reduced neointimal formation, further supporting the rational to use MMPi's in restenosis prevention.93,94 Although MMP inhibition was shown to be successful at inhibiting smooth muscle cell migration,95 in vivo studies have had mixed results in restenosis models.74,96,97 The BRILLIANT-EU study examined whether drug-eluting stents coated with the broad-spectrum MMPi, batimastat, would inhibit in-stent restenosis in patients without effects on re-endothelialization.98,99 The study concluded that batimastat-coated stents proved safe in larger populations (n = 550), although there was no net benefit at primary (major adverse cardiac events) or secondary (binary restenosis, subacute thrombosis, angiography) endpoints.97 Aside from their antibiotic effects, doxycycline and its derivatives have been shown to have broad-spectrum MMPi activity.97,100-102 A prospective study in 2004 (MIDAS)103 examined the effect of subantimicrobial doses of doxycycline in reducing the incidence of plaque rupture in acute coronary syndrome. Doxycycline-treated groups showed a 46% reduction in C-reactive protein levels and a 50% reduction in MMP-9 activity; however, there was no difference between treatment and placebo in the major cardiovascular endpoints, including myocardial infarction (MI) and death.103
The effect of inhibiting MMPs, including MMP-13, has also been examined in animal and human studies of ventricular remodeling after MI or cardiac injury. In the PREMIER phase 2 study, 253 patients with STEMI and poor ejection fraction were randomized to placebo or the MMPi, PG-116800,104,105 to examine the effects of broad-spectrum MMP inhibition on ventricular remodeling.106 PG-116800 was well tolerated but showed no significant improvements in left ventricular function at the 90 day endpoint. In experimental models of ventricular remodeling resulting from volume overload or heart failure, PD-166793, which blocks MMP-13, -2, and -3, showed significant protection against left ventricular dysfunction and detrimental remodeling in rats (Table 1).107-109 Another MMP-13 inhibitor (also blocks MMP-2, -3, -9) CP-471,474, showed significant reduction in left ventricular dilation after experimental MI in mice.110 These in vivo studies suggest that inhibiting various MMPs may provide beneficial effects; however, the contribution of specific inhibition of MMP-PAR1 signaling has not been fully assessed. Furthermore, achieving therapeutic levels of certain MMPi's in patients over long time periods has been limited by musculoskeletal toxicity (musculoskeletal syndrome) that has resulted in decreased dosing strategies and potential loss of efficacious MMPi levels.97 Patients with musculoskeletal syndrome experience joint pain, stiffness, and decreased mobility with continued MMPi treatment, which is not alleviated by nonsteroidal anti-inflammatory agents.97 Therefore, further development of orally active MMPi's with high specificity for the intended MMP target and identification of the detailed mechanism of inhibition may improve desired clinical outcomes.
In conclusion, MMPs have emerged as crucial mediators of platelet, endothelial, and vascular biology. Their traditional view as extracellular proteins with exclusively matrix-remodeling capabilities has shifted to a new focus on their roles as signaling molecules. Certain members of the interstitial collagenase family, such as MMP-1 and MMP-13, can directly activate G protein signaling through PAR1. MMP-1 and MMP-13 cleave the N-terminal extracellular domain of PAR1 at distinct sites from the thrombin cleavage site to generate unique tethered ligands, which activate biased signaling pathways. Exposure of platelets to collagen results in activation of MMP-1, which in turn directly cleaves PAR1 on the surface of platelets. Blocking the MMP1-PAR1 pathway inhibits early collagen-dependent thrombogenesis, arterial thrombosis, and clot retraction. Furthermore, in sepsis models, activation of the MMP1-PAR1 pathway leads to loss of vascular integrity and triggers systemic activation of inflammatory-coagulation cascade. MMP-13 was found to cleave and activate PAR1 on cardiac cells, which may be a new pathogenic pathway contributing to early heart failure. These findings suggest that the metalloprotease-PAR1 axis may present a new avenue for therapeutic intervention in multiple human diseases.
Presented in part at the Scientific Committee on Thrombosis and Vascular Biology at the 53rd Annual Meeting of the American Society of Hematology, San Diego, CA, December 10 and 11, 2011.
Acknowledgments
The authors thank Dr Andrew Bohm for generating structural models of MMP-1 and PAR1-derived ligands.
This work was supported in part by the National Institutes of Health (grant F30 HL108590, K.M.A.; grants R01 HL64701, RC2 HL101783, and P50 HL110789, A.K.; and grant R01 CA104406, L.C.) and United Against Lung (L.C.).
National Institutes of Health
Authorship
Contribution: K.M.A. and A.K. researched, wrote, and edited the manuscript; and L.C. researched and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Athan Kuliopulos, Laboratory of Hemostasis and Thrombosis, Molecular Oncology Research Institute, Tufts Medical Center, Box 7510, 750 Washington St, Boston, MA 02111; e-mail: athan.kuliopulos@tufts.edu.