

Abstract
Notch is a well-known oncogene in T-ALL, yet appears to have tumor suppressor effects in B-ALL. These cell type-specific effects of Notch signaling mirror consequences seen in early lymphocyte development and raises the question of how Notch leads to such divergent consequences in closely related cell types. In exploring these Notch mechanisms we discovered a B-ALL specific Notch-mediated reduction in the cell cycle regulator Polo-like kinase-1 (PLK1), revealing a novel targetable kinase in B-ALL.
To explore the consequences of Notch-mediated down regulation of cell cycle regulator kinase PLK1, we targeted PLK1 kinase function with the novel PLK1-selective inhibitor poloxin in human B-ALL lines.
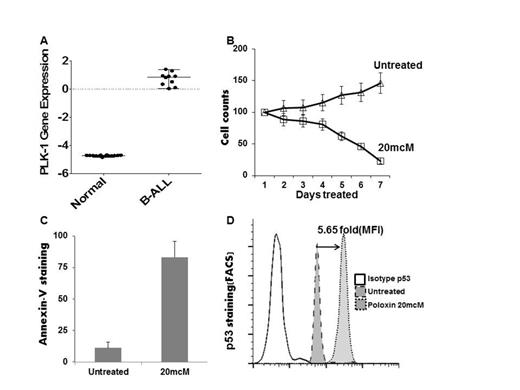
PLK1 is highly expressed in B-ALL verses normal tissues (panel A), correlates with cyclin B expression, is expressed >2-fold higher in B-ALL with t(1;19) than other B-ALL samples, and may predict response of ALL to methotrexate. In our panel of human B-ALL cell lines poloxin induced G2/M growth arrest and decreased cell number by >80% (panel B), and decreased survival in B-ALL cells (>75% AnnexinV+, panel C). PLK1 inhibition led to tumor suppressor p53 stabilization, revealing >5-fold increase in p53 protein levels following poloxin treatment in B-ALL (panel D). Mechanistically, PLK1 inhibition leads to both cytoplasmic re-localization of cyclin B, disrupting the CDC2-cyclinB complex, as well as phosphorylation of p53 at Ser20, which destabilizes p53-MDM2 interaction and thus accumulation of p53.
While exploring the mechanisms of cell type-specific effects of Notch signaling in ALL, we have found a novel therapeutic target, the cell cycle regulator PLK1. Our findings reveal a novel therapeutic approach whereby PLK1-selective inhibition via poloxin induces growth arrest and apoptosis in human B-ALL via consequences on cyclin B and p53 pathways.
No relevant conflicts of interest to declare.

