

Case Presentation

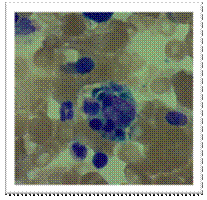
Bone marrow aspirate of this patient showing evidence of haemophagocytosis (H & E). A and B show large macrophages undergoing phagocytosis of different cellular elements of the bone marrow.
Bone marrow aspirate of this patient showing evidence of haemophagocytosis (H & E). A and B show large macrophages undergoing phagocytosis of different cellular elements of the bone marrow.
Several genes have been implicated in the genesis of primary HLH; all of them have in common impaired cytotoxic function by NK and T cells. It has been reported that mutations of the PRF1 gene comprise approximately 20 to 30% of the cases of primary HLH. This variant has been reported as a polymorphism and was not conclusive for the diagnosis of perforin deficiency as the cause of HLH in this case. In addition, the mutation was heterozygous. Although HLH is usually a recessive disorder, there are reports of potential disease associations in the heterozygous state. There is significant overlap between primary and secondary HLH and the role of some heterozygous mutations remain to be investigated.
No relevant conflicts of interest to declare.

