

In this issue of Blood, show that silent cerebral infarction (SCI) in sickle cell anemia occurs mainly in brain regions at the highest anatomic and hemodynamic vulnerability to ischemia, indicating that a tenuous and easily disrupted balance between oxygen supply and demand is the predominant cause of SCI.1
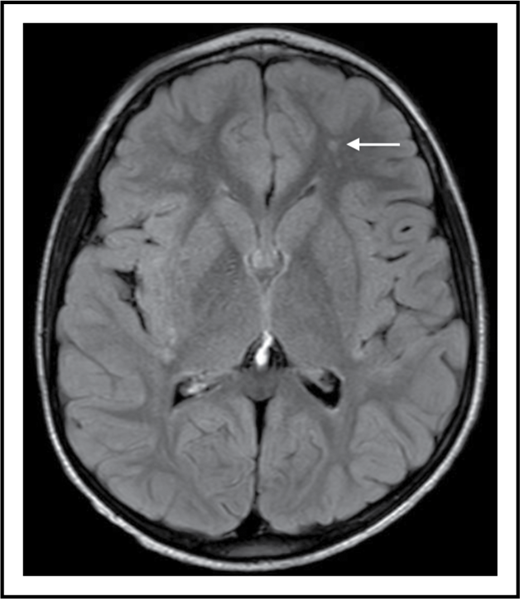
SCI is a supply-side problem in sickle cell anemia. The arrow shows a small stroke by magnetic resonance imaging (MRI) that has no corresponding deficit on physical examination (ie, SCI) in the left frontal white matter in a 6-year-old child with sickle cell anemia.
SCI is a supply-side problem in sickle cell anemia. The arrow shows a small stroke by magnetic resonance imaging (MRI) that has no corresponding deficit on physical examination (ie, SCI) in the left frontal white matter in a 6-year-old child with sickle cell anemia.
SCI is the term for a subset of strokes, usually small, that have no localizing signs. However, it should not be reassuring that the physical examination of an individual with SCI is normal, because the brain is composed of more than motor and somatosensory cortex and associated long tracts. These small strokes (see figure) are morbid, not silent, and can limit academic attainment and impair cognition in children and adults with sickle cell anemia.2 Moreover, SCI increases the risk for subsequent overt strokes, even in those who have normal transcranial Doppler (TCD) examinations.2,3
Because it is a covert lesion, which even neurologists cannot detect by examination,4 SCI must be identified by MRI scans of the brain. Like most organ injury in sickle cell anemia, SCI begins in early life. At a mean age of 14 months, 13% of infants who had MRI scans in the BABY HUG trial already had SCI.5 Thereafter, the prevalence of SCI increases with age, reaching 30% to 40% in adolescents and more than 50% in adults.2 Given that this progressive form of brain injury begins in early childhood, preventing SCI would be ideal. Primary prevention of SCI has not been established. However, DeBaun and colleagues3 demonstrated in the Silent Infarct Transfusion Trial (SIT Trial) that chronic transfusions can prevent recurrent cerebral infarction in children with SCI. In that trial, 196 children (age 5-15 years) with sickle cell anemia who had SCI and normal TCD examinations were randomly assigned to chronic transfusions or standard care (no transfusions) for 3 years. The relative risk reduction with chronic transfusions for recurrent infarction was 58% with a number needed to treat of 13.3
Why was transfusion therapy beneficial in the SIT Trial? To answer this question, one needs to understand the cause of SCI. Speculation has included vaso-occlusion in the brain, thrombosis, embolism, and small vessel disease (posited to be distinct from the occlusive cerebral vasculopathy that occurs in the large arteries around the circle of Willis). However, the main risk factors for SCI are lower baseline hemoglobin concentration (ie, more severe chronic anemia),6,7 acute anemic events,7,8 and higher systolic blood pressure.6 Together, these risk factors suggest a different cause for most SCI: a mismatch between oxygen supply and demand. To test this hypothesis, Ford and colleagues sought to demonstrate in children with sickle cell anemia that SCI occurs most commonly in regions of brain with the highest risk of ischemia, as defined both by anatomic vulnerability (occurrence in the anterior or posterior vascular border zones) and hemodynamic vulnerability (occurrence in regions with lowest measured resting cerebral blood flow [CBF]).
The investigators first outlined all the SCI lesions that were identified in the SIT Trial screening population (N = 286) and co-registered these to a standard brain atlas to create an SCI density map. They found evidence to support their hypothesis that most SCI lesions occurred in a small, vulnerable region of the brain. Specifically, 90% of SCI lesions were in vascular border zone distributions in deep white matter, which makes up only 5.6% of total brain volume. Resting CBF was then measured using arterial spin labeling in an independent group of children with sickle cell anemia (N = 41) with clinical characteristics similar to those in the SIT Trial screening population. As postulated, measured CBF was lowest in the independently defined regions of highest SCI density. In the ideal study, both of these measurements would be made in the same study population, but the expense and logistics of large-scale MRI studies make this a prohibitive design.
In summary, Ford and colleagues have shown that the majority of SCI occurs in regions of the brain that have the highest vulnerability to ischemia, by both anatomic and functional hemodynamic criteria. This speaks to an ongoing and tenuous balance between oxygen supply and demand in these brain regions in sickle cell anemia. These results explain why SCI occurs even during the overtly well or steady state9 and at an even higher frequency during acute exacerbations of anemia.8 The findings also demonstrate the pathophysiologic basis of known risk factors for SCI, including degree of chronic anemia, history of acute anemic events, and higher systolic blood pressure. These factors can dysregulate or decrease the supply of oxygen to the brain. Increased cerebral metabolic rate (increased oxygen demand), especially during acute illnesses, may further disrupt this tenuous balance. Therefore, one main mechanism by which chronic transfusions prevent SCI is alleviation of chronic anemia (and limitation of the degree of acute exacerbations of anemia), that is, improvement in the supply of oxygen to the brain.
These findings should also focus our attention and future studies on what we can do better to prevent SCI, such as disease-modifying therapies to decrease the degree of chronic anemia (as long as these interventions do not also impair oxygen delivery to the tissues). We also need to be aware that SCI can occur in the acute ischemic phase during acute anemic events.8 Because we cannot assess brain hemodynamics or detect SCI by physical examination,4 we should give strong consideration to transfusion for acute exacerbations of anemia, even when patients are hemodynamically stable and asymptomatic.
Finally, with the exception of vascular pruning, we should move away from the notion of SCI as a consequence of “small vessel disease” or, necessarily, proximal steno-occlusive cerebral vasculopathy. In the SIT Trial, fewer than 3% of the randomly assigned participants (with SCI and normal or conditional TCD measurements) had vasculopathy by magnetic resonance angiography, and progression of SCI was independent of abnormal TCD.10 The cause of most SCI seems to be the tenuous and easily disrupted balance between oxygen supply and demand in regions of brain most vulnerable to ischemia.
Conflict-of-interest disclosure: The author declares no competing financial interests.

