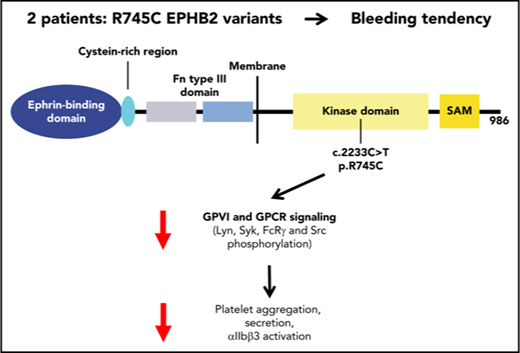
Key Points
EPHB2 tyrosine kinase receptor plays an important role in platelet function through crosstalk with GPVI and GPCR signaling.
A novel EPHB2 variant causes an inherited platelet function disorder.

Abstract
The ephrin transmembrane receptor family of tyrosine kinases is involved in platelet function. We report the first EPHB2 variant affecting platelets in 2 siblings (P1 and P2) from a consanguineous family with recurrent bleeding and normal platelet counts. Whole-exome sequencing identified a c.2233C>T variant (missense p.R745C) of the EPHB2 gene. P1 and P2 were homozygous for this variant, while their asymptomatic parents were heterozygous. The p.R745C variant within the tyrosine kinase domain was associated with defects in platelet aggregation, αIIbβ3 activation, and granule secretion induced by G-protein–coupled receptor (GPCR) agonists and convulxin, as well as in thrombus formation on collagen under flow. In contrast, clot retraction, flow-dependent platelet adhesion, and spreading on fibrinogen were only mildly affected, indicating limited effects on αIIbβ3 outside-in signaling. Most importantly, Lyn, Syk, and FcRγ phosphorylation, the initial steps in glycoprotein VI (GPVI) platelet signaling were drastically impaired in the absence of platelet–platelet contact, indicating a positive role for EPHB2 in GPVI activation. Likewise platelet activation by PAR4-AP showed defective Src activation, as opposed to normal protein kinase C activity and Ca2+ mobilization. Overexpression of wild-type and R745C EPHB2 variant in RBL-2H3 (rat basophilic leukemia) cells stably expressing human GPVI confirmed that EPHB2 R745C mutation impaired EPHB2 autophosphorylation but had no effect on ephrin ligand-induced EPHB2 clustering, suggesting it did not interfere with EPHB2-ephrin–mediated cell-to-cell contact. In conclusion, this novel inherited platelet disorder affecting EPHB2 demonstrates this tyrosine kinase receptor plays an important role in platelet function through crosstalk with GPVI and GPCR signaling.
Introduction
EPH receptors (erythropoietin-producing hepatoma-amplified sequence) are a large family of transmembrane tyrosine kinases that interact with ephrin ligands. These receptors, composed of an N-terminal glycosylated ligand-binding domain, a transmembrane region, and an intracellular kinase domain, are divided into 2 subclasses (A and B) based on sequence similarity and ligand affinity. Their ligands are also divided into 2 subclasses: the A subclass (A1-A6), which is tethered to the cell membrane by a glycosylphosphatidylinositol anchor, and the B subclass (B1-B3), which has a transmembrane domain followed by a short cytoplasmic region. Generally, ephrin A ligands bind to EPHA receptors, while ephrin B ligands bind to the EPHB receptors. One exception is EPHA4, which can bind both ephrins A and B. Ligand–receptor interactions initiate clustering, tyrosine phosphorylation, and signaling of EPHs.1,2 Cell surface clustering of the ligated ephrins elicits signaling in ephrin-bearing cells.3,4 EPH receptors and ephrins are expressed in virtually all tissues, and they are involved in several biological functions, including vascular development, tissue border formation, cell migration, axon guidance,2 memory learning,5 bone homeostasis,6 insulin secretion,7 and neurological development. These cellular responses to EPH-ephrin signaling also include changes in the actin cytoskeleton, cell-substrate adhesion, cell motility, and modulation of cell survival and proliferation. Finally EPH-ephrins are involved in various pathologies such as neurological disorders and cancer, in which EPHs and ephrins function during tumor invasion, neoangiogenesis, and metastasis.1,8 Indeed, several EPHs have been shown to exhibit tumor suppressor roles in prostate, colon, and breast cancer,9-11 prompting their exploration as potential therapeutic targets.
Human platelets are known to express EPHA4, EPHB1, and its ligand, ephrin B1.12-14 They promote platelet adhesion and aggregation as well as stability of the hemostatic plug, whereas inhibition of the EPH-ephrin interaction leads to disaggregation at low doses of agonists. EPHA4 and EPHB1 are also involved in adhesion to the fibrinogen matrix, triggering tyrosine phosphorylation of the β3 cytoplasmic domain of αIIbβ3- and αIIbβ3-mediated outside-in signaling controlling platelet spreading and platelet retraction. Recently, Vaiyapuri et al,15 using transgenic mice (EPHB2LacZ) expressing a chimeric EPHB2-β-galactosidase protein (with preserved ephrin binding but abrogated tyrosine kinase activity), concluded that EPHB2 was involved in the regulation of thrombus formation and retraction. Most importantly, they showed that the cytoplasmic tail of EPHB2 regulates platelet activation in a contact-independent manner in the absence of EPHB2-ephrin ligation between platelets.
We now report a new recessive bleeding disorder with impaired platelet function and a signaling defect that cosegregates with a predicted disease-causing EPHB2 gene variant affecting the EPHB2 receptor. The missense homozygous mutation p.R745C is located in the tyrosine kinase domain of EPHB2 and modifies a potential phosphorylation site. Both affected siblings from a consanguineous family were homozygous for the mutation. The defect gives impaired platelet aggregation and secretion induced by various agonists (thrombin, adenosine diphosphate [ADP], and convulxin [Cvx]) decreased adhesion and thrombus formation on collagen with modified glycoprotein VI (GPVI) and G-protein–coupled receptor (GPCR) signaling. Overexpression of wild-type (WT) and variant EPHB2 in RBL-2H3 (rat basophilic leukemia) cells stably expressing human GPVI suggests that EPHB2 positively regulates GPVI signaling.
Methods
Further details regarding the materials and methods used are provided in supplemental Methods (available on the Blood Web site).
Patients
Two patients, a brother (P1) and sister (P2) with an inherited bleeding syndrome, were the object of clinical and biological studies in the Bordeaux Reference Center for Platelet Pathologies prior to being enrolled within the BRIDGE-Bleeding and Platelet Diseases (BPD) cohort (UK REC10/H0304/66) (Cambridge, United Kingdom) after informed consent and in accordance with the Declaration of Helsinki. Studies were performed under the promotion of the Ethics Committee of Ile de France IV (IRB 00003835). Whole-exome sequencing (WES) identified a list of ∼200 variants for each patient that were then assessed taking into account the consanguinity of the family and their phenotype.
EPHB2 expression in the cell line
EPHB2-R745C complementary DNA (cDNA) was generated by site-directed mutagenesis of the full-length WT EPHB2 cDNA fused to the fluorescent reporter protein GFP and inserted in the pcDNA3 expression vector. WT or mutant EPHB2 cDNA was transiently transfected into RBL-2H3 cells16 by nucleofection using the Neon transfection system (Invitrogen/ThermoFisher Scientific, Saint-Aubin, France) according to the manufacturer’s instructions (1 µg plasmid DNA for 105 cells).
Statistical analysis
Results are presented as mean ± standard error of the mean (SEM). Data distribution was analyzed using the Student t test or 1-way analysis of variance followed by the Tukey multiple comparison test. A value of P < .05 was considered statistically significant (*P < .05, **P < .01, ***P < .001), and P > .05 was considered not significant.
Results
Patient characterization
The 2 affected patients are siblings of a consanguineous family (pedigree in Figure 1A). The brother (P1) and his sister (P2), now young adults, were respectively 12 and 15 years old when initial platelet investigations were performed (supplemental Table 1). In both cases, excessive spontaneous subcutaneous bleeding and heavy bleeding after minor wounds were observed. In addition, during childhood, P1 developed anemia resulting from chronic gastrointestinal bleeding requiring iron treatment. The International Society on Thrombosis and Hemostasis – Bleeding Assessment Tool bleeding score17 was 11 for P1 and 6 for P2. No other associated syndromes were observed. Both parents were asymptomatic. The platelet count was normal for all family members, but in a recent investigation (10 years later), a mild decrease in platelet count was noted for P1 (120 × 109 platelets/L). Other hematopoietic lineages were normal for all the family (supplemental Table 1). The initial platelet function investigations revealed a decreased response to ADP, arachidonic acid, collagen, and U46619 but a normal response to ristocetin (supplemental Table 1). Platelet responses were normal for both parents. Evaluation of platelet adenosine triphosphate (ATP)/ADP content using the luciferin-luciferase procedure and dense granule number evaluated by whole-mount electron microscopy revealed normal values for the patients P1 and P2 and their parents (supplemental Table 2). Other platelet function investigations detailed below were performed after the EPHB2 variant was selected as the primary disease-causing candidate.
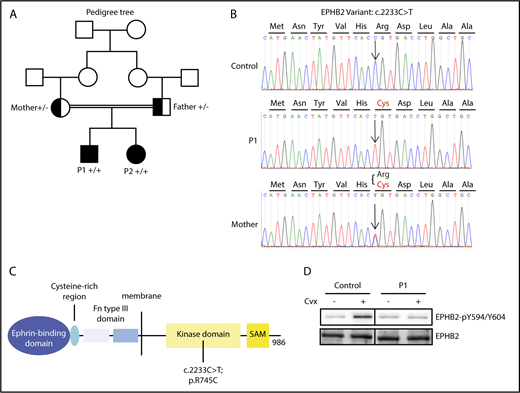
Identification of EPHB2 mutation. (A) The family tree (boxes, males; circles, females). The variant is indicated for P1 and P2 in solid black, while parents heterozygous for the mutation are indicated as half black box and circle (“+” signifies presence of the variant in one allele and “−” the WT in the other allele). (B) Sanger sequences surrounding the mutation (arrow) within the EPHB2 gene for patient P1 (upper sequence, homozygote), the mother (heterozygote), and an unaffected control. (C) Schematic representation of identified domains of the protein EPHB2. Fn, fibronectin; SAM, sterile α motif. (D) Phosphorylation of EPHB2 (EPHB2-pY594/604) in the presence or absence of Cvx (800 pM) was assessed in control and P1 platelets. Vertical lines have been inserted to indicate a repositioned gel lane.
Identification of EPHB2 mutation. (A) The family tree (boxes, males; circles, females). The variant is indicated for P1 and P2 in solid black, while parents heterozygous for the mutation are indicated as half black box and circle (“+” signifies presence of the variant in one allele and “−” the WT in the other allele). (B) Sanger sequences surrounding the mutation (arrow) within the EPHB2 gene for patient P1 (upper sequence, homozygote), the mother (heterozygote), and an unaffected control. (C) Schematic representation of identified domains of the protein EPHB2. Fn, fibronectin; SAM, sterile α motif. (D) Phosphorylation of EPHB2 (EPHB2-pY594/604) in the presence or absence of Cvx (800 pM) was assessed in control and P1 platelets. Vertical lines have been inserted to indicate a repositioned gel lane.
Genetic analysis
Six nonsynonymous homozygous rare variants shared by the 2 patients were identified by WES (supplemental Table 3) provided by BRIDGE-BPD, including p.R745C EPHB2. On the basis of the clinical and biological phenotype of both patients and similarities with the recent phenotype published for the β-Gal-EPHB2 transgenic mouse model,15 EPHB2 was selected as the most likely candidate gene. This gene, located on chromosome 1, encodes the EPHB2 receptor tyrosine kinase. The homozygous c.2233C>T transition was predicted to give rise to a p.R745C missense variant of the EPHB2 protein (transcript NM_004442.6). PolyPhen-2 software and the Sift score predicted the variant to be damaging with high probability. This EPHB2 variant was present in the ExAC database at minor allele frequency <10−6, showing that it is not a common polymorphism. Sanger sequencing of this variant in all individuals of the family confirmed that P1 and P2 were homozygous for the c.2233C>T transition, whereas the parents were heterozygous (Figure 1B). Interestingly, the variant amino acid is located in the tyrosine kinase intracytoplasmic domain of the molecule affecting one of the predicted tyrosine kinase phosphorylation sites (Figure 1C).18 Indeed, EPHB2 phosphorylation on Y594/604 (EPHB2-p-Y594/604) located in the juxtamembrane domain and required for kinase activity was increased in control platelets induced by Cvx (800 pM), while phosphorylation of tyrosines 594/604 was diminished in P1 platelets (Figure 1D). The tyrosine kinase domain including R745 is highly conserved among the different animal species from homo sapiens to fish (Danio rerio), suggesting a strong functional impact for the R745C mutation. Finally, R745 is located immediately before D746, the presumed proton acceptor essential for ATP-dependent kinase active site of the EPHB2 kinase domain. It is thus likely that changing the bulky basic R745 for the much smaller neutral C745 will interfere with D746 function.
None of the other 5 candidate rare variants were predicted to be disease causing, and no other candidate mutations were present in any gene known to be associated with an inherited platelet-derived bleeding disorder.
Morphological and structural characterization of platelets
Platelet count and volume for both patients were normal during adolescence, whereas the platelet count slightly decreased for patient P1 when he was 21 years old. Electron microscopy revealed heterogeneity in platelet shape; while some platelets appeared thin, discoid, and abnormally elongated, others were round (Figure 2A-F). A defect in fragmentation was suggested by platelets with a sickle shape and extensions of the plasma membrane (Figure 2A,F). Some large fragments of MK were present in the blood (Figure 2C,G), as were barbel-shaped preplatelets (Figure 2E). Quantitative morphometric evaluation of platelets of both patients confirmed a mean increase in maximal diameter (P1: 3.61 ± 0.84 μm, P ≤ .001; P2: 3.72 ± 0.83 μm, P ≤ .001 [vs control: 2.85 ± 0.15 μm]) whereas the minimal diameter was close to normal values (supplemental Table 4). As a consequence the ratio between maximal and minimal diameter was much higher (P1: 2.79 ± 1.22, P ≤ .1; P2: 3.27 ± 1.49, P ≤ .001 [vs control: 2.4 ± 0.1]). The large standard deviation reflects the heterogeneity. For patient P1 with the recent slight decrease in platelet count, the morphological platelet characteristics remain the same. Evaluation by manual counting of microtubule coils in electron microscopy photos revealed a similar number of microtubule coils for patient and control (control: 14.2 ± 4.6 microtubules; P1: 14.6 ± 4.4 microtubules; not depicted). Levels of EPHB2 (supplemental Figure 1A) and other membrane receptors, GPVI, FcRγ, β1, and αIIbβ3 (supplemental Figure 1B), α-granule contents (fibrinogen), α-granule/dense granule membrane marker (P-selectin), and β1-tubulin for microtubules (supplemental Figure 1C-E) were normal.
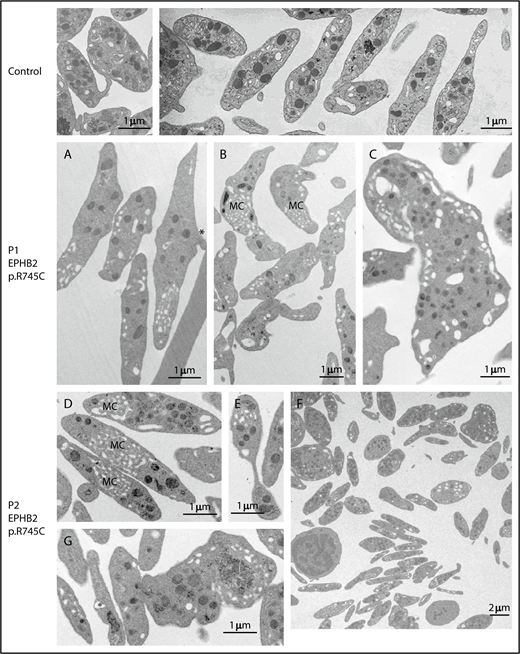
EPHB2 mutation affects platelet morphology. Illustration of the typical platelet ultrastructure for control (top), P1 (A-C), and P2 (D-G) by transmission electron microscopy. (A-B) Elongated platelets P1 with increased maximal diameter and a discoid shape containing few granules; note the abnormal presence of membrane complexes (MC). (A) Asterisk denotes examples of platelets with a sickle shape and extensions of the plasma membrane suggesting a defect of fragmentation. (C) What appears to be a fragment of a megakaryocyte (MK) with the aspect of an unfragmented proplatelet is shown. (D-G) Platelets from P2. (D) Discoid platelets with numerous membrane complexes. (E) An apparent preplatelet with a barbell-shaped appearance. (F) A lower-magnification image shows the heterogeneity in platelet shape; some are discoid and thin while others are round. (G) An unfragmented MK. Scale bars indicate the magnification.
EPHB2 mutation affects platelet morphology. Illustration of the typical platelet ultrastructure for control (top), P1 (A-C), and P2 (D-G) by transmission electron microscopy. (A-B) Elongated platelets P1 with increased maximal diameter and a discoid shape containing few granules; note the abnormal presence of membrane complexes (MC). (A) Asterisk denotes examples of platelets with a sickle shape and extensions of the plasma membrane suggesting a defect of fragmentation. (C) What appears to be a fragment of a megakaryocyte (MK) with the aspect of an unfragmented proplatelet is shown. (D-G) Platelets from P2. (D) Discoid platelets with numerous membrane complexes. (E) An apparent preplatelet with a barbell-shaped appearance. (F) A lower-magnification image shows the heterogeneity in platelet shape; some are discoid and thin while others are round. (G) An unfragmented MK. Scale bars indicate the magnification.
EPHB2 variant associates with decreased platelet aggregation and αIIbβ3 activation
Because initial platelet characterization showed abnormal platelet function, we looked in detail at platelet aggregation, secretion, and αIIbβ3 activation in both patients (P1 and P2). Aggregation of washed platelets induced by various concentrations of ADP (10 and 20 μM), thrombin (25 and 50 mU/mL), and PAR4-AP (50 μM) was strongly impaired (Figures 3A-C and 4A), but secretion was only partially affected (dense and α-granules) in both patients (Figures 3E-F and 4C). Since aggregation requires αIIbβ3 activation, we next quantified αIIbβ3 activation by binding of PAC1, an mAb specific for the activated conformation of human αIIbβ3 integrin. In the absence of stirring, αIIbβ3 activation induced by ADP, thrombin, or PAR4-AP was totally abolished in both patients (Figures 3B-D and 4B). We next investigated PAR4-AP–induced signaling after removal of secreted ADP by apyrase (2 U/mL) and inhibition of thromboxane A2 synthesis by indomethacin (10 μM). Because no Ca2+ mobilization was observed with PAR4-AP (50 μM) in unstirred control platelets (results not shown), higher doses of PAR4-AP (100 μM and 200 μM) were used (Figure 4D). Ca2+ mobilization and phosphorylation of protein kinase C (PKC) substrates were similar in P1 and controls, indicating that the activity of PLCβ3 was normal in P1 (Figure 4D-E). In contrast, no increase in PAR4-AP–induced Src activity, as assessed by the phosphorylation level of the amino acid Y418, was observed in P1 (Figure 4F), indicating that EPHB2 acts close to or directly on the tyrosine kinase Src and thus downstream of PLCβ3.
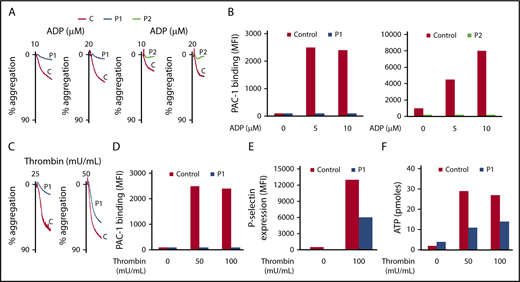
Platelet aggregation, secretion, and αIIbβ3 activation induced by ADP or thrombin are impaired in both patients. (A-C) Aggregation of washed platelets was initiated by various concentrations of ADP (10 and 20 μM) and thrombin (25 and 50 mU/mL) for 3 minutes. Aggregation was expressed as the percentage change in light transmission, with the value of the blank (buffer without platelets) set at 100%. Tracings are representative of at least 2 experiments per patient. (B-D) Quantification of activated αIIbβ3 at the surface of washed platelets was assessed by flow cytometry by binding of the specific monoclonal antibody (mAb) PAC1 to control and P1 and P2 platelets upon activation with ADP (5 and 10 μM) and thrombin (50 and 100 mU/mL). (E-F) Quantification of P-selectin exposure was assessed by flow cytometry using anti-P-selectin antibody and dense granule release with an ATP determination kit, respectively, to control and P1 platelets upon activation by thrombin (50 and 100 mU/mL). Results are representative of 2 experiments for each patient.
Platelet aggregation, secretion, and αIIbβ3 activation induced by ADP or thrombin are impaired in both patients. (A-C) Aggregation of washed platelets was initiated by various concentrations of ADP (10 and 20 μM) and thrombin (25 and 50 mU/mL) for 3 minutes. Aggregation was expressed as the percentage change in light transmission, with the value of the blank (buffer without platelets) set at 100%. Tracings are representative of at least 2 experiments per patient. (B-D) Quantification of activated αIIbβ3 at the surface of washed platelets was assessed by flow cytometry by binding of the specific monoclonal antibody (mAb) PAC1 to control and P1 and P2 platelets upon activation with ADP (5 and 10 μM) and thrombin (50 and 100 mU/mL). (E-F) Quantification of P-selectin exposure was assessed by flow cytometry using anti-P-selectin antibody and dense granule release with an ATP determination kit, respectively, to control and P1 platelets upon activation by thrombin (50 and 100 mU/mL). Results are representative of 2 experiments for each patient.

Downregulation of platelet signaling induced by PAR4-AP. (A) Aggregation of washed platelets for P1 and P2 was initiated by PAR4-AP (50 μM) for 3 minutes. Aggregation was expressed as the percentage change in light transmission, with the value of the blank (buffer without platelets) set at 100%. Tracings are representative of at least 2 experiments per patient. (B) Quantification of activated αIIbβ3 at the surface of washed platelets was assessed by flow cytometry by binding of the specific mAb PAC1 to control, P1, and P2 platelets upon activation by PAR4-AP (100 to 200 μM). (C) Quantification of P-selectin expression at the surface of washed platelets was assessed by flow cytometry by binding of the specific anti-P-selectin antibody to control, P1, and P2 platelets upon activation by PAR4-AP (100 to 200 μM). These results are representative of 2 experiments for each patient. (D) Ca2+ mobilization was assessed in unstirred platelets preincubated with the cytosolic Ca2+ fluorescent probe Oregon green BAPTA-AM after stimulation by PAR4-AP (100 and 200 μM) by flow cytometry in conditions of no external Ca2+ (1mM EGTA). (E-F) Phosphorylation of PKC substrates and Src-Y418 of control and P1 platelets activated by PAR4-AP (0 to 300 µM) was assessed by immunoblotting using an anti-PKC substrates (p-S) and anti-Src-p-Y418, respectively. Results are representative of 2 experiments. Quantification of Src phosphorylation (Src-p-Y418) was the average of the 2 experiments.
Downregulation of platelet signaling induced by PAR4-AP. (A) Aggregation of washed platelets for P1 and P2 was initiated by PAR4-AP (50 μM) for 3 minutes. Aggregation was expressed as the percentage change in light transmission, with the value of the blank (buffer without platelets) set at 100%. Tracings are representative of at least 2 experiments per patient. (B) Quantification of activated αIIbβ3 at the surface of washed platelets was assessed by flow cytometry by binding of the specific mAb PAC1 to control, P1, and P2 platelets upon activation by PAR4-AP (100 to 200 μM). (C) Quantification of P-selectin expression at the surface of washed platelets was assessed by flow cytometry by binding of the specific anti-P-selectin antibody to control, P1, and P2 platelets upon activation by PAR4-AP (100 to 200 μM). These results are representative of 2 experiments for each patient. (D) Ca2+ mobilization was assessed in unstirred platelets preincubated with the cytosolic Ca2+ fluorescent probe Oregon green BAPTA-AM after stimulation by PAR4-AP (100 and 200 μM) by flow cytometry in conditions of no external Ca2+ (1mM EGTA). (E-F) Phosphorylation of PKC substrates and Src-Y418 of control and P1 platelets activated by PAR4-AP (0 to 300 µM) was assessed by immunoblotting using an anti-PKC substrates (p-S) and anti-Src-p-Y418, respectively. Results are representative of 2 experiments. Quantification of Src phosphorylation (Src-p-Y418) was the average of the 2 experiments.
Since EPHB2 has been shown to be important for GPVI signaling,15 we next investigated Cvx-induced platelet aggregation. Cvx (200-800 pM)–induced platelet aggregation was totally abolished in both patients at low doses of Cvx (P1: 200 pM; P2: 200 and 400 pM) and drastically decreased at higher doses (Figure 5A). Similar results were obtained for collagen-induced platelet aggregation in platelet-rich plasma (supplemental Table 1). In parallel, PAC1 binding to αIIbβ3 in the patients’ platelets was completely abolished, regardless of the concentrations of Cvx used (200-800 pM) (Figure 5B). We next measured αIIbβ3 activation triggered by Cvx after removal of ADP by apyrase (2 U/mL) and inhibition of thromboxane A2 synthesis by indomethacin (10 μM). The partial αIIbβ3 activation observed in control platelets under these conditions was therefore directly dependent on GPVI (Figure 5C). The total absence of αIIbβ3 activation observed in patients’ platelets under the same conditions (apyrase and indomethacin) showed that the EPHB2 mutation directly affected GPVI signaling. Altogether, these results demonstrate that the EPHB2 mutation affects platelet aggregation and αIIbβ3 activation in a manner both dependent and independent of GPVI signaling.
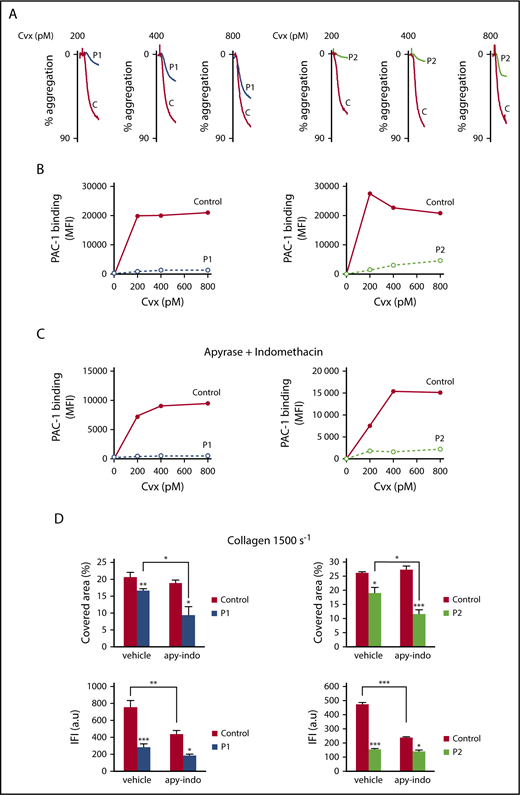
Platelet aggregation and αIIbβ3 activation induced by Cvx are altered in both patients. (A) Aggregation of washed platelets was initiated by increasing concentrations of Cvx (200-800 pM) for 3 minutes. Aggregation was expressed as the percentage change in light transmission, with the value of the blank (buffer without platelets) set at 100%. Tracings are representative of at least 2 experiments per patient. (B) Quantification of activated αIIbβ3 at the surface of washed platelets was assessed by flow cytometry by binding of the mAb PAC1 to control and P1 and P2 platelets upon activation with Cvx (200-800 pM). (C) The same experiment was conducted in the presence of apyrase (2 U/mL) and indomethacin (10 μM). Results are representative of 2 experiments for each patient. (D) Adherence and thrombus size were investigated on collagen matrix (50 μg/mL) in the presence or absence of apyrase (2 U/mL) and indomethacin (10 μM) at high shear rate (1500 s−1) after 3 minutes. Platelet adhesion was expressed as covered surface (%) and thrombus size as total integrated fluorescence intensity (IFI) from 2 experiments carried out in triplicate. Data are presented as the mean ± SEM and statistical differences were determined using 1-way analysis of variance followed by the Tukey multiple comparison test (*P < .05, **P < .01, ***P < .001). a.u, arbitrary units.
Platelet aggregation and αIIbβ3 activation induced by Cvx are altered in both patients. (A) Aggregation of washed platelets was initiated by increasing concentrations of Cvx (200-800 pM) for 3 minutes. Aggregation was expressed as the percentage change in light transmission, with the value of the blank (buffer without platelets) set at 100%. Tracings are representative of at least 2 experiments per patient. (B) Quantification of activated αIIbβ3 at the surface of washed platelets was assessed by flow cytometry by binding of the mAb PAC1 to control and P1 and P2 platelets upon activation with Cvx (200-800 pM). (C) The same experiment was conducted in the presence of apyrase (2 U/mL) and indomethacin (10 μM). Results are representative of 2 experiments for each patient. (D) Adherence and thrombus size were investigated on collagen matrix (50 μg/mL) in the presence or absence of apyrase (2 U/mL) and indomethacin (10 μM) at high shear rate (1500 s−1) after 3 minutes. Platelet adhesion was expressed as covered surface (%) and thrombus size as total integrated fluorescence intensity (IFI) from 2 experiments carried out in triplicate. Data are presented as the mean ± SEM and statistical differences were determined using 1-way analysis of variance followed by the Tukey multiple comparison test (*P < .05, **P < .01, ***P < .001). a.u, arbitrary units.
Cvx-induced platelet secretion and thrombus formation on collagen matrix are impaired in patients with the EPHB2 variant
We next quantified dense and α-granule secretion by measuring ATP release and P-selectin exposure. The total content of α-granule (fibrinogen) (supplemental Figure 1C) and dense granule secretion (ATP) was checked and confirmed to be normal (results not shown). In Cvx-induced aggregation, ATP release was decreased by 60% to 70% for P1 platelets and 44% to 100% for P2 platelets compared with control platelets (supplemental Figure 2A). In unstirred conditions, P-selectin exposure at a low dose of Cvx (200 pM) was 71% and 74% lower in P1 and P2 platelets than in control platelets (supplemental Figure 2B). The addition of apyrase (2 U/mL) and indomethacin (10 μM) clearly showed that the decrease in P-selectin exposure is largely dependent on GPVI signaling in P1 and P2 (supplemental Figure 2C). Only a small part of P-selectin exposure was dependent on ADP release and TXA2 synthesis under unstirred conditions (supplemental Figure 2B-C).
We next assessed the impact of this variant on adhesion and thrombus formation on a collagen matrix at arterial shear rate (1500 s−1). For these experiments, platelet counts were normalized. The area covered by P1 and P2 platelets was slightly lower, reaching 80.5% and 72.5% of controls (100%) (P1: P < .01; P2: P < .05) (Figure 5D, top). Thrombus size was significantly decreased for P1 and P2, reaching 37.6% for P1 (P < .001) and 32.2% for P2 (P < .001) (Figure 5D, bottom). On addition of apyrase and indomethacin, the residual adhesive platelets and thrombus size were significantly lower in P1 (P < .05) and in P2 (P < .001) than in controls (Figure 5D); thus, the decreases for the patients’ platelets were both dependent and independent of GPVI signaling. Altogether, these results indicate that the EPHB2 mutation affects secretion and GPVI signaling involved in thrombus formation.
EPHB2 variant only mildly affects αIIbβ3 outside-in signaling
Because EPHB2 has been reported to be physically associated with αIIbβ3 in mouse platelets and involved in the regulation of the outside-in signaling,15 we next investigated the impact of this EPHB2 variant on αIIbβ3-mediated outside-in signaling. First, the kinetics of clot retraction was analyzed in platelet-rich plasma after the addition of various concentrations of thrombin (0.1 and 0.5 U/mL). A reduction of clot retraction induced by 0.1 U/mL thrombin was already observed at 20 minutes, reaching 53.8% and 38.2% of control values for P1 and P2; however, after 1 hour of thrombin treatment (0.1 U/mL) or at a higher concentration of thrombin (0.5 U/mL), clot retraction normalized (supplemental Figure 3A). Because the lower rate of clot retraction may be explained by a defective response to thrombin (thrombin-induced platelet aggregation and αIIbβ3 activation were partly decreased in P1 and P2; Figure 3C), we next examined adhesion to fibrinogen matrix (100 µg/mL) under low shear conditions (300 s−1) and in the presence of apyrase/indomethacin. Adhesion was moderately but significantly reduced, reaching 79% (P < .01) and 67% (P < .01) of controls (100%) for P1 and P2, respectively (supplemental Figure 3B). Note that spreading on a fibrinogen matrix (100 µg/mL) under static conditions was also moderately decreased (78.5% of control) (supplemental Figure 3C). Altogether, these results indicate that the EPHB2 variant only slightly alters platelet αIIbβ3 outside-in signaling.
GPVI signaling pathway is impaired in the patients’ platelets
To get insight into the mechanism of the regulation of platelet function by EPHB2, we next examined the signaling pathway downstream of GPVI using unstirred platelets in the presence of apyrase/indomethacin, preventing superimposed signaling by ADP or thromboxane A2. First we assessed Ca2+ mobilization from intracellular stores, which is essential for platelet activation and secretion. Flow cytometry of Oregon green BAPTA1-AM–loaded platelets stimulated with Cvx (200-800 pM) showed a lower Ca2+ mobilization for P1 (Figure 6A) and P2 (supplemental Figure 4), regardless of the concentration of agonist used. The activation of phosphatidylinositol 3-kinase and PLCγ2, important players in GPVI signaling, as assessed by the phosphorylation level of its effector Akt (Akt: p-S473) and PLCγ2 (PLCγ2-p-1217) were totally or partially abolished for P1 (Figure 6B). We next examined the initial steps of GPVI signaling by measuring the phosphorylation of the tyrosine phosphorylation of the immunoreceptor tyrosine-based activation motif motif of FcRγ (FcRγ-P) and the tyrosine kinase Syk (Syk: p-Y525/526) associated with GPVI. Syk-P and FcRγ-P as detected by anti-Syk-P and anti-phosphotyrosine antibodies, respectively, were also drastically impaired in P1 platelets (Figure 6C), strongly suggesting that EPHB2 regulates the first steps of the GPVI signaling pathway. Since the activation of Src kinases (Lyn and Fyn), which are constitutively associated with GPVI, plays a critical role in initiating GPVI signaling by mediating the phosphorylation of FcRγ and Syk on tyrosine residues, we next examined the phosphorylation of Lyn. Lyn contains 2 highly conserved tyrosine residues: a C-terminal one exhibiting an inhibitory activity (Lyn: p-Y507), and another one that is part of the activation loop (Lyn p-Tyr 396).19 Under our conditions, as expected, Lyn activation (Lyn p-Y396) was increased in control platelets after Cvx stimulation (Figure 6D). Conversely, no increased phosphorylation of Lyn p-Tyr 396 after Cvx stimulation was observed in P1 platelets. Altogether, these results confirm that EPHB2 acts during the initial step of GPVI signaling and independently of platelet–platelet contact.
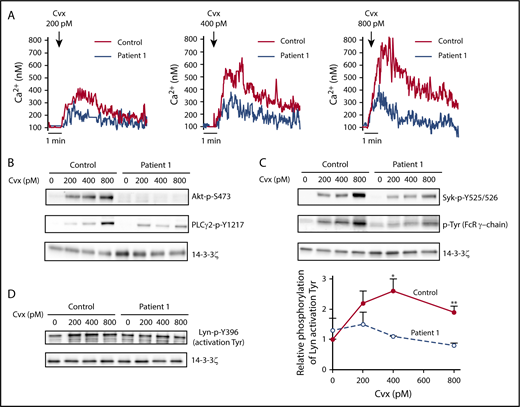
Downregulation of platelet signaling induced by Cvx. Washed P1 platelets in suspension were activated by Cvx (200 or 800 pM) for 3 minutes in the absence of stirring and in the presence of apyrase (2 U/mL) and indomethacin (10 μM). (A) Ca2+ mobilization was assessed in unstirred platelets preincubated with the cytosolic Ca2+ fluorescent probe Oregon green BAPTA-AM after stimulation by Cvx (200-800 pM) by flow cytometry in conditions of no external Ca2+ (1 mM EGTA). (B-C) Tyrosine phosphorylation of Akt (p-S473), Syk (p-Y525/526), PLCγ2 (p-Y1217), and FcRγ was assessed by immunoblotting using anti-Akt-P, anti-Syk-P, anti-PLCγ2-P, and anti-phosphotyrosine antibodies, respectively. Results are representative of 2 experiments. (D) Lyn phosphorylation on Y396 was assessed by immunoblotting using an anti-Lyn-P. Data represent mean ± SEM of 4 independent experiments (*P < .05, **P < .01, unpaired Student t test).
Downregulation of platelet signaling induced by Cvx. Washed P1 platelets in suspension were activated by Cvx (200 or 800 pM) for 3 minutes in the absence of stirring and in the presence of apyrase (2 U/mL) and indomethacin (10 μM). (A) Ca2+ mobilization was assessed in unstirred platelets preincubated with the cytosolic Ca2+ fluorescent probe Oregon green BAPTA-AM after stimulation by Cvx (200-800 pM) by flow cytometry in conditions of no external Ca2+ (1 mM EGTA). (B-C) Tyrosine phosphorylation of Akt (p-S473), Syk (p-Y525/526), PLCγ2 (p-Y1217), and FcRγ was assessed by immunoblotting using anti-Akt-P, anti-Syk-P, anti-PLCγ2-P, and anti-phosphotyrosine antibodies, respectively. Results are representative of 2 experiments. (D) Lyn phosphorylation on Y396 was assessed by immunoblotting using an anti-Lyn-P. Data represent mean ± SEM of 4 independent experiments (*P < .05, **P < .01, unpaired Student t test).
Confirmation of the effect of EPHB2 R745C mutation on EPHB2 function by overexpression in GPVI-expressing RBL-2H3 cells
One of the key features of EPH receptors is that they undergo autophosphorylation upon crosslinking, which is usually mediated by cell–cell contact and ephrin engagement. To test whether EPHB2 R745C mutation affected autophosphorylation in the absence of cell–cell contact, we made use of the RBL-2H3 cell line stably expressing recombinant human GPVI in which WT- and EPHB2-variant constructs were overexpressed. The efficiency of transfection was between 30% and 40% for EPHB2-WT and the EPHB2-variant. In the absence of Cvx, no phosphorylation of EPHB2 was observed in WT and mutant EPHB2 (Figure 7A). RBL-2H3 cell activation by Cvx (1 nM) led to phosphorylation of WT and mutant EPHB2, but to a lesser extent for the latter (50% of WT-EPHB2), indicating that GPVI activation can activate EPHB2 in the absence of ephrin–EPHB2 interaction.
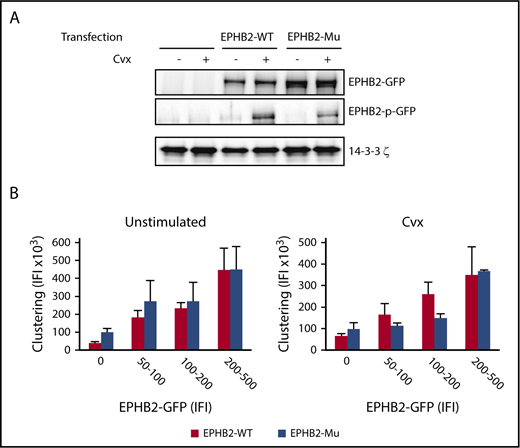
Effect of EPHB2/p.R745C mutation on autophosphorylation of EPHB2 and ephrin-induced clustering. RBL-2H3 cells transfected with WT and mutant EPHB2 constructs were plated on fibrinogen coverslips for 1 hour. (A) Overexpression and phosphorylation of EPHB2-WT and EPHB2/p.R745C (EPHB2-μ) variant after activation with Cvx (1 nM) was quantified by western blotting using anti-EPHB2 and EPHB2-pY594/Y604 antibodies. Results are representative of 3 independent experiments. (B) Clustering of EPHB2 was performed by adding ephrin B1-Fc dimeric ligand to transfected cells stimulated by Cvx (1 nM) and plated on fibrinogen-coated coverslips for 30 minutes. After various washes, binding of ephrin B1-Fc and the level of EPHB2-GFP for the corresponding cell were quantified by fluorescence microscopy (Nikon Eclipse E600). Total IFI from 3 independent experiments is presented as the mean ± SEM.
Effect of EPHB2/p.R745C mutation on autophosphorylation of EPHB2 and ephrin-induced clustering. RBL-2H3 cells transfected with WT and mutant EPHB2 constructs were plated on fibrinogen coverslips for 1 hour. (A) Overexpression and phosphorylation of EPHB2-WT and EPHB2/p.R745C (EPHB2-μ) variant after activation with Cvx (1 nM) was quantified by western blotting using anti-EPHB2 and EPHB2-pY594/Y604 antibodies. Results are representative of 3 independent experiments. (B) Clustering of EPHB2 was performed by adding ephrin B1-Fc dimeric ligand to transfected cells stimulated by Cvx (1 nM) and plated on fibrinogen-coated coverslips for 30 minutes. After various washes, binding of ephrin B1-Fc and the level of EPHB2-GFP for the corresponding cell were quantified by fluorescence microscopy (Nikon Eclipse E600). Total IFI from 3 independent experiments is presented as the mean ± SEM.
For the patients, we found that the abnormal platelet response was not dependent on cell–cell contact, thus excluding a role for the ephrin ligands EPHB2-GPVI crosstalk. To confirm these results, we tested the capacity of EPHB2 to form clusters in the presence of an ephrin-Fc ligand in cell line considering that EPHB2 clustering by ephrin mimics EPHB2 response to cell–cell contact. First, WT-EPHB2 clustering increased with the level of recombinant WT-EPHB2 in RBL-2H3 cells (Figure 7B). No statistical difference was seen for the EPHB2 mutant in the presence or absence of Cvx regardless of the level of GFP-WT and GFP-EPHB2 variant overexpressed (Figure 7B). Altogether, these results indicate that the EPHB2/p.R745C variant interferes with the first steps of cell activation in a contact-independent manner but has no direct functional effect in conditions of cell–cell contact.
Discussion
The evidence suggesting that EPHB2 plays a major role in platelet function is growing. Here, we identified a new inherited platelet bleeding disorder affecting the EPHB2 gene in 2 siblings from a consanguineous family. Both are homozygous for the missense EPHB2/p.R745C variant, whereas the asymptomatic parents are heterozygous for this variant. Both patients exhibited recurrent bleeding and normal or slightly decreased platelet counts. Platelet functions (aggregation, secretion induced by different agonists, and thrombus formation on a collagen matrix) and αIIbβ3 activation were severely impaired. In contrast, clot retraction and adhesion on a fibrinogen matrix under flowing blood were only moderately affected. This indicates that the EPHB2 variant mostly affects αIIbβ3 inside-out rather than outside-in signaling.
This variant identified by WES was selected among a group of homozygous variants common to both siblings and in an heterozygous state in both parents (supplemental Table 3) on the basis of similarities between patients’ platelet phenotype and platelet defects reported in transgenic mice expressing EPHB2 with the cytoplasmic domain replaced by β-galactosidase.15 Few inherited platelet function disorders affecting several agonists have been described, the most common being Glanzmann thrombasthenia and, more recently, Ca2+ and DAG-regulated guanine nucleotide exchange factor I–related disorder,20 a severe bleeding disorder resulting from a signaling protein defect primarily involving the inside-out activation of αIIbβ3.21,22 The abnormalities for EPHB2 were not limited to platelet function and also concerned platelet morphology, with occasional thin and abnormally long platelets completely different from the large platelets that characterize macrothrombocytopenia (MYH9-RD, FLNA-RD, BSS,and VWD2B), where the majority of platelets are spheroid and of large size with an increased small diameter.23 Nonetheless, this observation suggests a role for EPHB2 in platelet biogenesis.
The defect in platelet functional responses to several agonists was particularly evident for collagen and Cvx. In the absence of platelet-to-platelet contact, proximal GPVI signaling in the patients’ EPHB2 was characterized by an impaired phosphorylation of Lyn, FcRγ, and Syk, suggesting that EPHB2 acts directly on GPVI and independently of its ligand, ephrin (that is dependent upon signaling processes involving direct contact between cells). GPVI activation triggered autophosphorylation of both WT and EPHB2/p.R745C, but to a lesser extent for the latter, suggesting impaired kinase activity. Interestingly, it has been shown that the autophosphorylation site of EPHB2 was the predominant binding site for the tyrosine kinase Src.24 Thus, a direct or indirect association of EPHB2 with Src kinases (Fyn and Lyn) constitutively bound to GPVI is an attractive possibility and should be tested. In good agreement with our observation, platelets from transgenic mice for β-galactosidase-EPHB2 displayed diminished phosphorylation of Akt and PLCγ2, similar to what we observed in the patients’ platelets. Surprisingly phosphorylation of Src, Lyn, and Syk was normal in EPHB2-β-galactosidase mice but low in our patient. The reason for this difference is unclear, and may lie in the difference between the deletion of (or its swapping for β-galactosidase) the entire cytoplasmic tail of EPHB2 and the Arg/Cys missense mutation, or in species differences.
Another important question is: what happens when ephrin binds to EPHB2/p.R745C, a condition encountered in platelet aggregation? Indeed, the interaction of EPH and ephrin B1, the only ephrin identified in platelets, has been shown to contribute to sustained platelet aggregation.14 The clustering of EPHB2/p.R745C, quantified after addition of ephrin B1, was not significantly affected; consequently, we speculate that the p.R745C variant does not affect EPHB2-ephrin B1 ligand–mediated platelet–platelet contact and thus thrombus stability. These data are in agreement with the EPHB2-β-Gal transgenic mouse model, where replacement of EPHB2 cytoplasmic tail by β-galactosidase, does not diminish clustering and signaling through ephrin B1.15 The impaired platelet aggregation was essentially the result of the absence of αIIbβ3 activation due to impaired GPVI proximal signaling.
While an inherited GPVI defect leads only to a mild bleeding phenotype,25 both of our patients with the EPHB2 variant exhibit a severe bleeding phenotype, strongly suggesting that EPHB2 is involved in other regulatory platelet or vascular functions. Most likely, this is because thrombin and ADP pathways are also strongly impaired, as shown on our aggregation and secretion studies with these agonists. Moreover, whereas PAR4-AP–induced PKC activity and Ca2+ mobilization were normal in P1, Src kinase activity was inhibited, indicating that EPHB2 plays a relevant role at a distal level of G-coupled receptors. The fact that EPHB2/p.R745C affects the phosphorylation of Lyn and Src induced, respectively, by Cvx and PAR4-AP, suggests that this EPHB2/p.R745C inhibits phosphatases involved in the dephosphorylation of inhibitory tyrosine residues of Src required for its activation.19
In summary, we describe a new inherited platelet disorder of a EPHB2/p.R745C variant characterized by a bleeding syndrome, abnormal elongated platelets, and a severe platelet function defect. Importantly, this EPHB2 variant alters GPVI signaling at the level of the GPVI receptor in the absence of cell–cell contact excluding involvement of ephrin binding during the initial platelet activation and GPCR signaling. Despite documented EPHB2 expression in many cell types, the absence of associated clinical signs (other than the bleeding syndrome) in the patients suggests that the effects of this variant are essentially restricted to the megakaryocyte-platelet lineage, most likely because of the absence of compensatory mechanisms (such as other EPH receptors) in this lineage present in other tissues.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients who participated to this study. They also thank Alice Davy and Thomas Jungas from the University of Toulouse and Nicolas Prévost from the University of Kyoto for fruitful discussions, Bruno Copin (engineer in bioinformatics) from Trousseau Hospital, and Christelle Repérant from U1176 INSERM for technical assistance.
This work was supported by INSERM.
Authorship
Contribution: E.B., C.S., F.A., R.F., A.K., Z.E., M.F., K.D., A.M., J.-C.B., P.B., S.L., and M.B. performed experiments; P.N., R.F., and S.G. provided clinical data and enrolled the patients; E.T. performed the bioinformatic analysis of DNA sequencing data; E.B., J.-P.R., P.N., and M.B. analyzed results; E.B., J.-P.R., and M.B. designed experiments; E.B., P.N., J.-P.R., and M.B wrote the manuscript; and E.B., C.S., R.F., F.A., A.K., A.T.N., J.-C.B., E.B., W.H.O., C.V.D., M.J.-P., and J.-P.R. critically edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Marijke Bryckaert, INSERM UMR S 1176, Hôpital Bicêtre, 80 rue du Général Leclerc, 94276 Le Kremlin Bicêtre Cedex, France; e-mail: marijke.bryckaert@inserm.fr.