Abstract
Blastoid mantle cell lymphoma is characterized by highly aggressive features and a dismal clinical course. These blastoid and pleomorphic variants are defined by cytomorphological features, but the criteria are somewhat subjective. The diagnosis may be supported by a high cell proliferation based on the Ki-67 labeling index. Recent analyses have shown that the Ki-67 index overrules the prognostic information derived from the cytology subtypes. Nevertheless, genetic analysis suggests that blastoid and pleomorphic variants are distinct from classical mantle cell lymphoma. In clinical cohorts, the frequency of these subsets varies widely but probably represents ∼10% of all cases. Chemotherapy regimens commonly used in mantle cell lymphoma, such as bendamustine, rarely achieve prolonged remissions when given at the dosage developed for classical variants of the disease. Thus, high-dose cytarabine–containing regimens with high-dose consolidation may be generally recommended based on the more aggressive clinical course in these patients. However, even with these intensified regimens, the long-term outcome seems to be impaired. Thus, especially in this patient subset, allogeneic transplantation may be discussed at an early time point in disease management. Accordingly, targeted approaches are warranted in these patients, but clinical data are scarce. Ibrutinib treatment results in high rates of responses, but the median duration of remission is <6 months. Similarly, lenalidomide and temsirolimus result in only short-term remissions. Novel approaches, such as chimeric antigenic receptor T cells, may have the potential to finally improve the dismal long-term outcome of these patients.
The histological concept of blastoid MCL
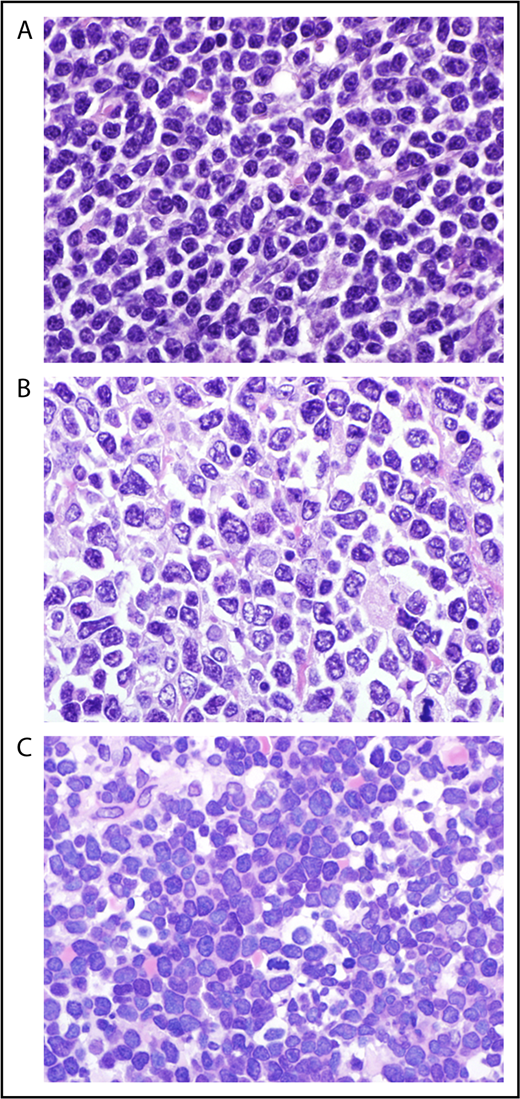
The term “blastoid mantle cell lymphoma” (MCL) describes a morphological subgroup of lymphomas with blastic features. Two commonly distinguished cytological variants share blastic morphology and high proliferation, the pleomorphic and blastoid variants.1 The lymphoma cells in the pleomorphic variant are larger than in conventional MCL. Their nuclear shape and chromatic structure resemble diffuse large B-cell lymphoma to some extent (Figure 1). The blastic variant morphologically resembles lymphoblasts found in lymphoblastic lymphoma/leukemia, with roundish nuclei, a narrow rim of cytoplasm, and finely dispersed chromatin (Figure 1).
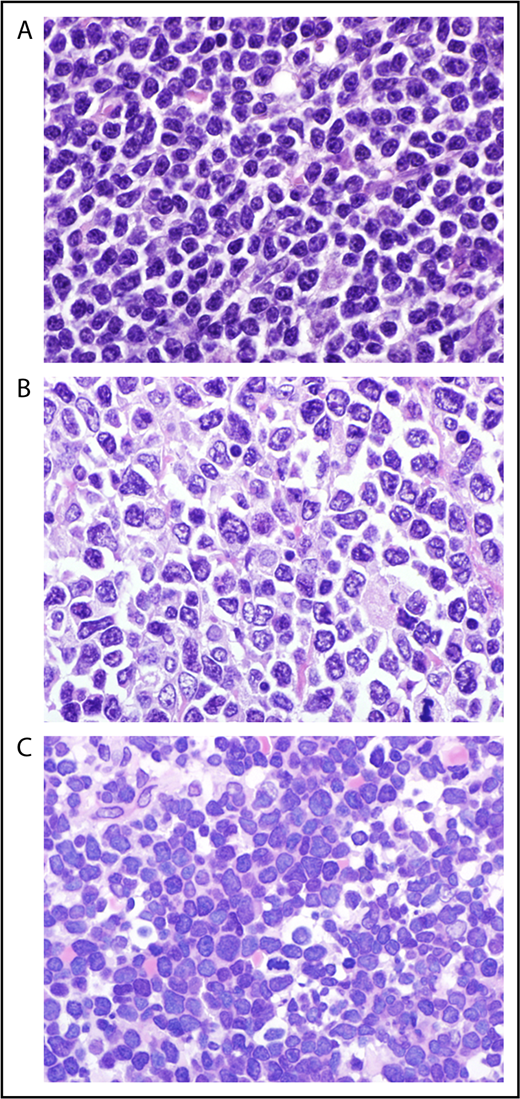
Cytology of mantle cell lymphoma classical (A), pleomorphic (B), and blastoid (C) MCL (original magnification ×1000, hematoxylin and eosin stain).
Cytology of mantle cell lymphoma classical (A), pleomorphic (B), and blastoid (C) MCL (original magnification ×1000, hematoxylin and eosin stain).
Common to the pleomorphic and blastic variants is the high proliferation index and the unfavorable outcome.2 However, the World Health Organization (WHO) classification suggests distinguishing the 2 variants.3 The definition is based on the morphology only. Accordingly, exclusive detection of high proliferation, as defined by the Ki-67 index, is not sufficient to classify as the blastoid or pleomorphic subtype, because classical MCL might also show high cell proliferation.4,5 However, the proliferation index certainly influences a pathologist’s decision to assign a cytology subtype to a lymphoma. This might explain, at least in part, the variability in the frequency of the blastoid and pleomorphic subtypes, which are reported to range from 10%6 to >20%2 of all MCL. Another feature that might explain differences in the frequencies of the blastoid and pleomorphic subtypes is the lack of guidelines on how to classify MCL that harbors lymphoma cells with a blastoid and classical cytology.7 We recommend reporting the cytology features and classifying these lymphomas as the blastoid or pleomorphic subtype.
Histological features
Blastoid morphology is predominantly a feature of MCL at first diagnosis6 ; however, in a subset of MCL, the morphology might switch from classical to blastoid during the course of the disease.8,9 An inverse pattern of evolution may even be observed in rare cases.9 However, studies analyzing sequential biopsies in MCL are exceedingly rare, and the true incidence of transformation remains uncertain.9 Blastoid morphology is associated with certain histological and immunophenotypical features. These features are only variably associated with the cytology subtype and do not substitute the cytological assessment. The growth pattern of blastoid variants is usually diffuse, less frequently is nodular, and rarely exhibits a mantle zone pattern. An “in situ” pattern of blastoid MCL has not been observed.
Immunophenotypic variability has been reported for blastoid MCL. Lack of expression of CD5 seems to occur more frequently than in classical subtypes.10 CD20 and Cyclin D1 seem to almost always be positive. Blastoid morphology is associated with a high proliferation, as measured by the Ki-67 index (see “Clinical presentation and prognostic significance”).7
TP53 protein overexpression, as detected by immunohistochemistry, seems to correlate well with the mutational status of the gene, whereas homozygous deletions of the locus are rarely detected in cases with no11 protein expression. Interestingly, TP53 overexpression, detected by immunohistochemistry and indicating genomic mutations, represents a much more dominant prognostic factor than genomic deletions only.12,13
Blastoid MCLs are characterized by a high level of c-MYC expression, despite the fact that c-myc translocations are rare.14 Nevertheless c-myc amplifications have been reported to occur frequently in blastoid MCL,15 and translocations of MYC have been reported in this subtype of MCL.16 Studies analyzing the microenvironment of blastoid MCL are rare; however, the presence of blastoid histology seems to be associated with a reduced number (W.K., unpublished observations) and a more diffuse pattern of follicular dendritic cells within the tumor tissue.17
Genetic alterations
It is important to note that pathognomonic genetic alterations that exclusively characterize blastoid variants, but are absent in classical MCL, have not been described. In fact, most molecular features are enriched among blastoid MCL but may also be detected in highly proliferative classical MCL. Genomic alterations found in blastoid variants might even precede the “transformation” from a classical to a blastoid variant.18 Like most MCLs, blastoid variants harbor the translocation t(11;14)(q13;q32) and consequently overexpress cyclin D1 messenger RNA and protein. The level of cyclin D1 RNA in MCL is higher when the 3′-untranslated region is deleted, a feature that is frequently found in blastoid variants of MCL.19,20 The blastoid variants show a level of chromosomal aberrations that even exceeds the high levels found in classical MCL.21 The pathways affected by the chromosomal gains and losses predominantly affect mechanisms of cell cycle control. Frequent secondary genomic alterations are deletions and losses of p16INK4a and p21Waf1 genes,22,23 as well as mutations of ATM and TP53.11 As a consequence, high levels of TP53 protein expression are detectable in a subset of blastoid MCL (see above).11 In fact, alterations in TP53 seem to be the chromosomal aberration that is most closely correlated, although not exclusively associated, with blastoid morphology.18 Systematic genomic analysis of MCL also revealed several recurrently mutated genes, including drivers of the cell cycle, such as ATM, cyclin D1, TP53, antiapoptotic protein BIRC3 and Toll-like receptor, ubiquitin ligase UBR5, and chromatin modifiers (WHSC1, MLL2, MEF2B).24,25 Among these genes, recurrently mutated Notch1 and Notch2 have been reported to be enriched among patients with blastoid morphology.25,26 Based on pathological observations and genetic studies, a model of MCL pathogenesis and progression toward blastoid variants has been suggested. This model assumes that increasing genetic alterations are leading to the loss of cell cycle control, the higher proliferation rate, and blastoid features.27 However, it has to be noted that blastoid features are frequently seen at initial presentation in some patients, whereas other cases remain morphologically stable classical MCL throughout the course of the disease.9,28,29 Most of the genetic alterations described seem to occur in blastoid and pleomorphic MCL. It remains uncertain which features might be unique to either subtype; however, pleomorphic variants have been shown to be more frequently tetraploid MCL than blastoid MCL.30
Clinical presentation and prognostic significance
The clinical presentation of blastoid disease resembles that of classical MCL, although systematic comparisons are scarce. Thus, the disease predominantly affects male patients who are diagnosed with advanced stage disease in their sixth decade of life, and some analyses report a slightly younger age than for classical MCL.31,32 Although initially suggested as a poor prognostic marker, predominant gastrointestinal involvement usually represents a low-proliferation more-indolent type of MCL. Clinically, a more aggressive clinical course of the blastoid variant may be observed based on the high cell proliferation of the malignant cells. A large analysis of the trials of the European MCL Network confirmed that the Ki-67 index overrides the prognostic significance of the cytology subtype in multivariate analyses.6 In addition, assessing TP53 protein expression and/or mutational analysis has become a promising prognostic tool.24,33 Thus, the value of cytology subtypes as prognostic subgroups of MCL has become less important. Nevertheless, we still consider the assessment of the cytology subtypes as a standard parameter to be assessed in any pathology report and clinical trial as an auxiliary tool to identify patients at high risk for relapse and progression.
Conventional chemotherapy
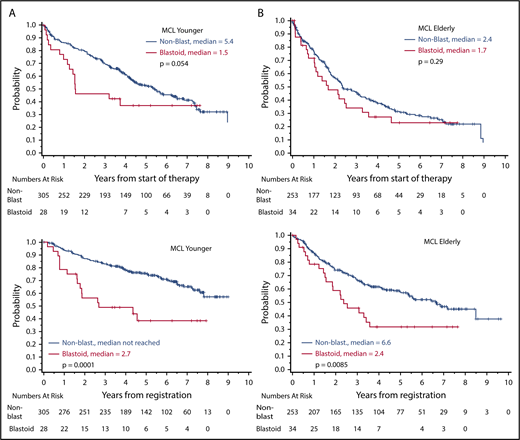
In historical series, blastoid morphology represents 1 of the most prominent prognostic factors in MCL.7 In the prerituximab era, complete response (CR) rates for blastoid variants were 36%, with a median response duration of 11 months only after conventional dosed chemotherapy (mostly CVP [cyclophosphamide, vincristine, prednisone] or CHOP [cyclophosphamide, doxorubicine, vincristine, prednisone]).32 Accordingly, the median overall survival (OS) was only 14.5 months in comparison with 53 months in 154 patients with classical MCL. The addition of rituximab has improved response rates and time to treatment failure (TTF) in a randomized trial and a meta-analysis, but data on blastoid variants are not available.34,35 Similarly, rituximab maintenance prolongs progression-free survival (PFS) after combined immunochemotherapy, but blastoid cytology remains a poor prognostic factor (Figure 2; Table 1).6,36
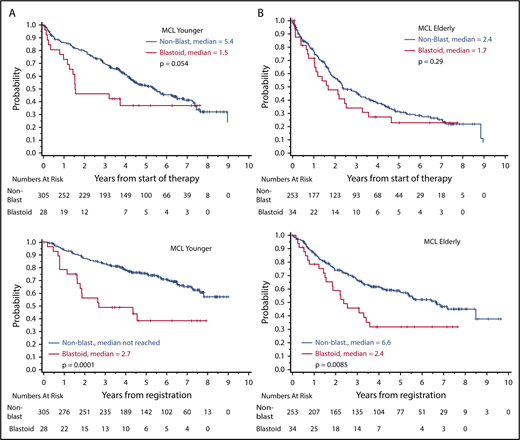
Overall survival according to cytology (blastoid variant vs classical MCL). (A) MCL younger: younger patients (n = 325) received R-CHOP with or without R-DHAP (rituximab, dexamethasone, high dose cytarabine, cisplatinum), followed by autologous stem cell transplantation. (B) MCL elderly: older patients (n = 295) were treated with R-CHOP vs R-FC (rituximab, fludarabine, cyclophosphamide), followed by interferon/rituximab maintenance.
Overall survival according to cytology (blastoid variant vs classical MCL). (A) MCL younger: younger patients (n = 325) received R-CHOP with or without R-DHAP (rituximab, dexamethasone, high dose cytarabine, cisplatinum), followed by autologous stem cell transplantation. (B) MCL elderly: older patients (n = 295) were treated with R-CHOP vs R-FC (rituximab, fludarabine, cyclophosphamide), followed by interferon/rituximab maintenance.
No major data set is available for bendamustine-based inductions; however, based on personal experience, blastoid variants are either refractory or tend to relapse rapidly after completion of treatment, but they can be salvaged by cytarabine-containing regimens. Therefore, an Italian study group has explored high-dose cytarabine in combination with bendamustine and rituximab in relapsed disease and as first-line treatment.37,38 Hematological toxicity (ie, thrombocytopenia) was major, with grade 3/4 in 87% of patients. However, 90% of 40 patients responded (CR, 83%). In contrast, at least in relapsed disease, the majority of blastoid variants relapsed within 9 months in comparison with almost 80% ongoing remissions in classical MCL. To improve tolerability, especially in elderly patients, a reduced-dose regimen has been explored as first-line treatment.38 Unfortunately, in the majority of cases, the small subset of patients with the blastoid variant (n = 6) relapsed within 1 year. Alternatively, an alternating regimen displayed a more favorable toxicity profile.39
The European MCL Network is performing a randomized trial investigating alternating rituximab + CHOP (R-CHOP)/R–high-dose cytarabine induction. This trial will address the efficacy of high-dose cytarabine in elderly patients with blastoid morphology.
Dose-intensified cytarabine-containing regimens
Because of the poor prognosis, it seemed intuitive to introduce dose-intensified concepts, especially in this patient subset. Based on the observation that cytarabine-based regimens are active in highly proliferative leukemias, various groups have implemented such concepts.40-46 In fact, in a historical comparison of trials, the clinical outcome of blastoid MCL seems to have benefited from the dose-intensive approaches, with or without autologous transplantation (Table 1).
The MD Anderson group reported an impressive 97% overall response (87% CR) with the hyper-CVAD (cyclophosphamide, vincristine, doxorubicine, dexamethasone) regimen plus rituximab in 97 patients with MCL.40 The 14 patients with the blastoid variant achieved a median TTF of 6.8 years, but this slightly inferior outcome was not significant in a multivariate analysis including the MCL international prognostic index (MIPI) and β2-microglobulin. In a historical comparison, a Scandinavian group confirmed the benefit of cytarabine and rituximab-containing induction, followed by autologous transplantation, for all subtypes of MCL after a median follow-up of 11.4 years.41,42 In a univariate analysis, blastoid histology resulted in an inferior OS (51% vs 61%; P = .018), and a trend toward a shorter response duration was observed (48% vs 57%; P = .055). In a multivariate analysis, cytology was displaced by the closely associated, but more reliably measured, cell proliferation (Ki-67 ≥ 30%) for event-free survival ([hazard ratio] HR, 2.32; P = .001) and OS (HR, 2.64; P = .001). However, high-dose cytarabine alone is not able to overcome the poor clinical course in high-risk cases. The Scandinavian Nordic group had initiated a further dose-intensified protocol (6 cycles of high-dose cytarabine 3 g/m2 every 12 hours, days 1 and 2) in high-risk MCL primarily characterized by high cell proliferation, but the study was stopped early because of insufficient responses (4 failures in the 5 initial patients).43 Interestingly, all 4 patients were salvaged by R-maxi-CHOP. A similar observation has been reported for the French LyMa trial, supporting the statement that anthracyclines may still add to the efficacy of cytarabine-containing regimens in high-risk MCL.44
The CALGB has investigated a similar dose-intensified regimen.45 Interestingly, this approach remained effective, even after reducing the methotrexate dose significantly, from 3 g/m2 to 300 mg/m2, because of serious renal toxicity in 8 of the initial 20 patients. Therefore, given the potential toxicity of methotrexate, most current regimens avoid this drug. For patients with blastoid MCL (n = 12), PFS was 56% and OS was 64% at 5 years.
Finally, in a randomized comparison of 497 MCL patients, the addition of cytarabine led to a significant improvement in TTF in the overall group (65% vs 40%; HR, 0.56; P = .038), and a similar value was detected in the subset with Ki-67 ≥ 30% (HR, 0.54); however, the blastoid variants again performed significantly worse than did classical MCL (5-year OS, 38% vs 75%; P = .0001; Figure 2).46
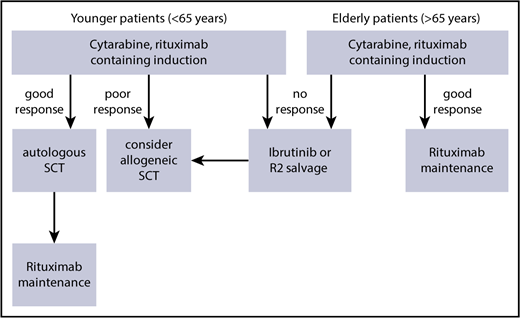
In summary, the outcome of blastoid MCL seems to have improved significantly with dose-intensified strategies in younger patients (Table 1); however, the outcome of blastoid variants remained significantly worse than that of classical MCL6 (Figure 2). Thus, in the event of an insufficient response, we recommend moving quickly to alternative approaches (Figure 3).
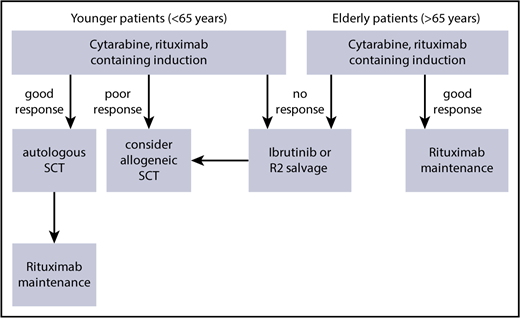
Suggested therapeutic algorithm in high-risk MCL (blastoid, Ki-67 > 30%, Tp53 mutation).
Suggested therapeutic algorithm in high-risk MCL (blastoid, Ki-67 > 30%, Tp53 mutation).
CNS involvement
In various series, blastoid cytology predisposes for central nervous system (CNS) involvement.47 In a retrospective survey by the European MCL Network, 57 of 1396 patients (4.1%) had CNS involvement, including 0.9% at diagnosis.48 Blastoid variants had a significantly higher rate of CNS involvement (28% vs 10%; P < .0001). Additional risk factors included B-symptoms, increased serum lactate dehydrogenase, poor Eastern Cooperative Oncology Group performance status, and high MIPI score. Similarly, in a Japanese study, 33 of 608 MCL patients (5.4%) were diagnosed with CNS involvement.49 Again, univariate risk factors included blastoid variant (HR, 3.11; P = .0006) and leukemic presentation, high MIPI score, and high cell proliferation (Ki-67). In a multivariate analysis, Ki-67 ≥ 30% remained the only independent risk factor (HR, 6.03; P = .003), probably substituting for blastoid histology due to the more reliable dichotomic differentiation. Thus, CNS prophylaxis may be considered, especially in the blastoid variant of MCL, although data on the efficacy of intrathecal prophylaxis or systemic treatment with methotrexate or high-dose cytarabine are inconclusive.47-50
Novel agents
With respect to the activity of novel agents in patients with blastoid-variant MCL, the evidence is somewhat sparse. In general, trials with novel agents are relatively small, and blastoid patients are not specifically identified, or they are excluded as part of the trial design. Despite this, the current literature gives a good impression of the general activity in this challenging group of patients.
Ibrutinib
There is the greatest amount of available data for ibrutinib. In the original phase 2 trial,51 111 patients were included, of which 17 had blastoid histology. The overall response rate was 68%; however, in the subset analysis, the presence of the blastoid variant did not appear to affect the outcomes. In a randomized trial of ibrutinib vs temsirolimus,52 12% of patients in both arms had a blastoid histology. Within the trial, blastoid cytology was identified as a significant poor prognostic factor (P < .05); no obvious differences were observed between the 2 arms, but numbers were very small. Outcomes of these patients could only be reliably analyzed after data from 3 trials with single-agent ibrutinib in MCL were pooled together.53 This analysis included 370 patients, of which 12% had blastoid MCL. When looking specifically at the difference between this group and the nonblastoid patients, the time to best response was similar (2.2 vs 2.1 months); however, the overall response rate was lower (55% vs 72%), the duration of the response was shorter (8.6 vs 18.8 months), and PFS (5.1 vs 14.6 months) and OS (12.8 months vs not reached) were inferior. Taken together, it appears that responses are significantly worse in blastoid MCL, suggesting that, although this agent is highly active in this context, it is likely that different approaches, including combination therapies, may be more appropriate.
The only published combination data including such patients involves the use of ibrutinib with rituximab. In this small trial of 50 patients with relapsed MCL,54 the responses and quality of responses were improved over single-agent ibrutinib. Interestingly, however, the overall response rate was different when the proliferation fraction was considered. Of the patients with Ki-67 < 50%, the response rate was 100%, in contrast to the 12 patients with Ki-67 > 50%, for whom the response rate was only 50%, and 2 of the nonresponding patients were noted to have blastoid histology. Therefore, although the addition of rituximab generally improves responses in MCL, it does not appear to be sufficient to overcome the poorer outcome seen with the more proliferative tumors, which largely represents those with the blastoid histology.
A further piece of evidence suggesting that the blastoid patients have inferior outcomes with ibrutinib comes from the 2 studies that described the very poor outcomes in patients relapsing following ibrutinib.55,56 These 2 articles describe the outcomes of the first cohort of patients progressing under ibrutinib. In the first,55 32% of patients described had blastoid histology prior to the initiation of therapy, which increased slightly (to 36%) at relapse. This percentage is much higher than one would expect, which reflects the fact that this group of patients relapsed early on therapy and, therefore, were overrepresented in this early analysis. In the second larger multicenter study of 114 patients,56 18 patients were known to have blastoid histology; in addition, 72% of patients with available data had Ki-67% ≥ 30%. Although the outcomes of these groups were not specifically analyzed, the patients have far more proliferative disease than would be expected in a relapsed refractory cohort, again suggesting that the earliest progressions on this agent were observed in the more proliferative cases and likely derived the shortest benefit from the drug.
In summary, although ibrutinib is extremely active as a single-agent in the context of MCL, including patients with the blastoid subtype, responses are generally inferior inpatients with the more proliferative type of disease. Different strategies are likely needed that probably should involve the addition of other agents, including chemotherapy.
Lenalidomide
Lenalidomide is also widely used; however, in the 2 largest studies, the pivotal MCL-001 (EMERGE) trial57 and the large randomized trial of lenalidomide vs investigator choice (SPRINT),58 there is no mention of the blastoid variant. The addition of rituximab to lenalidomide has been widely applied in MCL; however, in the relapse study59 of 52 patients, there is no mention of the blastoid variant, and, it is specifically stated that no such patients were included in the front-line trial.60 In fact, it appears that the more proliferative tumors tend to do worse with lenalidomide. A subsequent analysis of EMERGE trial data evaluated outcomes with respect to proliferation, as assessed by Ki-67 fraction.61 Although response rates were the same when a cutoff of 30% was applied, PFS (1.9 vs 4.5 months) and OS (9.7 and 28.4 months) were significantly worse for those patients with a higher proliferative fraction. This was slightly worse when a cutoff of 50% was applied for PFS (1.8 vs 4.5 months) and OS (7.4 vs 23.9 months). It seems likely that any blastoid patients would be represented within these more proliferative cohorts and, by inference, have a worse outcome. The European MCL Network is performing a randomized trial investigating alternating R-CHOP/R–high-dose cytarabine induction, followed by rituximab with or without lenalidomide maintenance. This trial will address the efficacy of lenalidomide maintenance in elderly patients with blastoid morphology.
Interestingly, a recently reported series of relapsed MCL suggests that the combination of ibrutinib and lenalidomide may overcome the negative prognostic impact of TP53 that is closely correlated with blastoid MCL.62 Response rates (TP53mut, 89% vs TP53wild-type, 88%) and PFS after 6 months were comparable. However, because of the small numbers and limited follow-up, these data are only hypothesis generating so far.
Bortezomib
The registration trials for bortezomib (as monotherapy63 or in combination with a CHOP-like regimen64 ) do not list the results of blastoid patients. However, in a small number of patients with high proliferation fraction, the addition of this agent prolonged the median PFS, with a similar HR as in the total study population.
Temsirolimus
Immunologic approaches
Approaches investigated in clinical trials include the application of PD-1 inhibitors, bispecific antibodies, and chimeric antigen receptor T cells. In general, published data for MCL are very scarce. However, the potential curative approach of allogeneic transplantation in blastoid variants supports the potential efficacy of immunological strategies. In a small subset of 4 patients with relapsed MCL, no response to nivolumab monotherapy has been observed, but blastoid cytology has not been reported.66 In contrast, a CD5/cD19 T-cell–engaging bispecific antibody has achieved objective responses in 5 of 7 patients with relapsed MCL.67,68 Single responses have also been reported for autologous chimeric antigen receptor T cells, in a very limited data set, by various investigators.69-71
In summary, limited data suggest that proliferation also remains an important determinant of response with novel agents when given by themselves or in combination with rituximab. Because they are highly proliferative tumors by inference, this would apply to the blastoid variants. As such, combination approaches will need exploration in this group; therefore, this specific patient cohort should not be excluded from new trials.
Conclusions
With regard to diagnosis and therapeutic strategies, blastoid MCL remains a challenge. In general, cases are identified by high cell proliferation (Ki-67 ≥ 30%) and numerous genetic alterations; TP53 mutations, especially, seem to represent a critical prognostic marker.
Based on the aggressive clinical course, a watch-and-wait strategy is not recommend, even in cases with low tumor burden. Instead, cytarabine-containing regimens, with or without autologous stem cell consolidation, should be preferred but have to be balanced against the expected therapy-associated toxicity.
Even with such an optimized therapeutic approach, blastoid MCL displays an inferior long-term outcome. Thus, an allogeneic approach may be discussed during the early course of the disease (Figure 3). However, for the majority of (elderly) patients, new strategies, including molecular approaches targeting the B-cell receptor pathway or immunological approaches, are being tested in clinical trials. Likewise, only combined approaches using these therapeutic tools will be able to overcome the otherwise dismal long-term prognosis for blastoid MCL.
Authorship
Contribution: All authors participated in writing the article.
Conflict-of-interest disclosure: M.D. received (institutional) research support from Celgene, Janssen, Mundipharma, and Roche and honoraria for giving presentations and serving on the scientific advisory boards for Bayer, Celgene, Gilead, Janssen, Mundipharma, Roche, and Sandoz. W.K. has no competing financial interests. S.R. received (institutional) research support from Janssen and honoraria for serving on the scientific advisory boards for AstraZeneca, Celgene, Celltrion, Gilead, Janssen, Napp, Pharmacyclics, Roche, and Sunesis.
Correspondence: Martin Dreyling, Department of Medicine III, University Hospital/LMU Munich, Marchioninistrasse 15, D-81377 Munich, Germany; e-mail: martin.dreyling@med.uni-muenchen.de.