Key Points
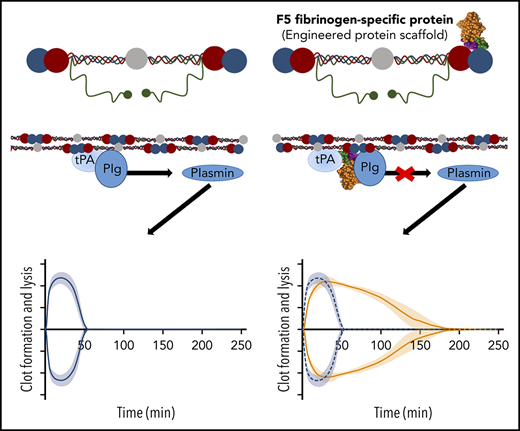
We describe a fibrinogen-specific conformational protein (Affimer) that delays fibrinolysis by reducing fibrin-dependent plasmin generation

Abstract
Bleeding complications secondary to surgery, trauma, or coagulation disorders are important causes of morbidity and mortality. Although fibrin sealants are considered to minimize blood loss, this is not widely adopted because of its high cost and/or risk for infection. We present a novel methodology employing nonantibody fibrinogen-binding proteins, termed Affimers, to stabilize fibrin networks with the potential to control excessive bleeding. Two fibrinogen-specific Affimer proteins, F5 and G2, were identified and characterized for their effects on clot structure/fibrinolysis, using turbidimetric and permeation analyses and confocal and electron microscopy. Binding studies and molecular modeling identified interaction sites, whereas plasmin generation assays determined effects on plasminogen activation. In human plasma, F5 and G2 prolonged clot lysis time from 9.8 ± 1.1 minutes in the absence of Affimers to 172.6 ± 7.4 and more than 180 minutes (P < .0001), respectively, and from 7.6 ± 0.2 to 28.7 ± 5.8 (P < .05) and 149.3 ± 9.7 (P < .0001) minutes in clots made from purified fibrinogen. Prolongation in fibrinolysis was consistent across plasma samples from healthy control patients and individuals at high bleeding risk. F5 and G2 had a differential effect on clot structure and G2 profoundly altered fibrin fiber arrangement, whereas F5 maintained physiological clot structure. Affimer F5 reduced fibrin-dependent plasmin generation and was predicted to bind fibrinogen D fragment close to tissue plasminogen activator (tPA; residues γ312-324) and plasminogen (α148-160) binding sites, thus interfering with tPA–plasminogen interaction and representing 1 potential mechanism for modulation of fibrinolysis. Our Affimer proteins provide a novel methodology for stabilizing fibrin networks with potential future clinical implications to reduce bleeding risk.
Introduction
Blood loss and treatment modalities that aim to replace blood components after traumatic vessel injury represent an important cause of morbidity and mortality.1,2 After surgical or accidental trauma, both the cellular and protein arms of coagulation are activated to form a hemostatic plug to stop blood loss. The blood clot is composed of a network of fibrin fibers with platelets, red blood cells, and other blood elements embedded in this complex.3 After vascular injury, soluble plasma fibrinogen is converted into an insoluble fibrin network, which is further stabilized by factor XIII (FXIII) that crosslinks fibrin fibers and incorporates antifibrinolytic proteins into the clot, limiting fibrinolysis and preventing excessive blood loss.4,5
Developing new therapies that minimize blood loss during acute bleeding episodes arising from trauma, or those associated with some hematological disorders, will help improve clinical outcome. The fibrin network has been the target of such therapies, and a number of fibrin sealants have been developed.6-9 Fibrin sealants are usually composed of a protein/reagents mixture including fibrinogen, FXIII, thrombin, and antifibrinolytic agents (aprotinin, tranexamic acid). This approach is promising in a variety of surgical procedures10-13 ; however, current fibrin sealants have a number of limitations including cost, anaphylactic reactions, and the risk for intravascular coagulation if inadvertently injected into the circulation. Furthermore, commercial preparations made from human plasma carry the risk for infection. Fibrin sealants made from recombinant proteins have been described but are yet to become widely available.14 Given these drawbacks, the clinical use of fibrin sealants has been limited.
Modulating fibrinolysis to stabilize the fibrin clot has been used for decades to limit blood loss. The widely used tranexamic acid inhibits plasmin activity, but the effects are partial and with a modest clinical benefit.15-17 The use of FXIII-mimetics to stabilize the blood clot and reduce fibrinolysis is a promising approach, but is yet to be tested in human studies.18
We developed a novel system that employs small proteins, termed Affimers,19,20 that have the capability to perform conformational interactions and are easily produced recombinantly in large quantities.19,21-23 We hypothesized that fibrinogen-specific Affimer proteins can be used to stabilize fibrin networks and increase resistance to lysis, with the potential to reduce bleeding in high-risk conditions. Therefore, our aims were to isolate fibrinogen-binding Affimer reagents, study modulation of fibrin clot lysis by fibrinogen-specific Affimer proteins, and explore mechanistic pathways for changes in fibrinolysis, if any, including alterations in fibrin clot structural properties.
Materials and methods
Screening for fibrinogen-binding Affimer proteins
Commercially available human fibrinogen (Calbiochem, Feltham, United Kingdom) was further purified, using IF-1 monoclonal antibody (Kamiya Biomedical, Seattle, WA), as described,24 to obtain highly pure protein. IF-1 purified fibrinogen was then biotinylated using EZ-Link NHS-SS-Biotin (Thermo Fisher Scientific, Loughborough, United Kingdom). After confirming the biotinylated fibrinogen remained functional in a turbidimetric assay (detailed here), it was added to streptavidin-coated wells (Thermo Fisher Scientific), followed by the addition of the Affimer phage library. The full screening protocol has been described previously.25
Affimer proteins of interest were produced by subcloning the coding regions into pET11a vector and expressing in BL21 (DE3) Escherichia coli cells (Thermo Fisher Scientific). All Affimer proteins contained a C-terminal polyhistidine (His)-tag and were purified using Ni-NTA sepharose (IBA, Loughborough, United Kingdom), as previously described.19
Synthesis of linear peptides
Four linear peptides of the same sequence as the variable regions of Affimer proteins F5 (P1F5 and P2F5) and G2 (P1G2 and P2G2) were synthesized by Thermo Fisher Scientific with peptide purity higher than 95%, followed by testing in the turbidimetric assay.
Turbidimetric assays
Whole-blood lysis
Samples of free-flowing blood were collected from the antecubital vein of 3 healthy volunteers into 0.109 M sodium citrate. Informed written consent was obtained from each volunteer in accordance with the declaration of Helsinki. Ethical approval was obtained from the University of Leeds Medical School Ethical Committee. Blood samples were recalcified and mixed with Affimer protein diluted in saline solution (0.9% NaCl), and 2.5 nM tissue plasminogen activator (tPA). Clotting was initiated with ex-tem reagent (Werfen, Warrington, United Kingdom) and followed for up to 5 hours at 37°C. Measurements were performed on a ROTEM δ and raw data extracted using Export Tool ROTEM δ V1.3. Lysis time was calculated from time of maximum clot firmness (maximum amplitude reached during clotting) to time of 50% reduction in maximum clot firmness.
Laser scanning confocal microscopy and scanning electron microscopy
Plasma clots were prepared in the presence of increasing concentrations of Affimer proteins, and visualized as previously described (details in supplemental Methods).26
Clot permeation
This was performed as previously described28 ; full details can be found in supplemental Methods.
Binding of Affimer proteins to fibrinogen
Enzyme-linked immunosorbent assay-based binding assay
Nunc-Immuno MicroWell 96 well-plates (Thermo Scientific) were coated with 5 µg/mL fibrinogen (Calbiochem, Feltham, United Kingdom) in 50 mM sodium carbonate at pH 9.6. A dilution series of Affimer protein F5 (0-2000 nM) or G2 (0-100 nM) was added to the wells. Mouse anti-His antibody (Roche, Welwyn Garden City, United Kingdom) and horseradish peroxidase-conjugated rabbit anti-mouse antibody (Dako, Glostrup, Denmark) were used to detect the Affimer proteins (details in supplemental Methods).
Competitive binding assays were performed to investigate tPA and plasminogen binding to fibrin DD fragment in the presence of Affimer protein F5. In addition, Affimer F5 was screened by enzyme-linked immunosorbent assay (ELISA) for nonspecific binding to tPA and plasminogen (experimental details can be found in the supplemental Methods).
Biacore surface plasmon resonance (SPR) binding assay
IF-1 purified human fibrinogen (Calbiochem; 5 µg/mL in 0.1 M sodium acetate buffer at pH 5.6) was immobilized to 2000 RUs by amine-coupling using an N-hydroxysuccinimide/(N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide–activated CM5 chip, followed by deactivation with ethanolamine/HCl, using a Biacore 3000 (GE Healthcare, Buckinghamshire, United Kingdom). An additional fibrin surface was prepared and binding assays were performed after a modified protocol from Ajjan et al29 (full details in supplemental Methods).
Pulldown assays for identification of F5–fibrinogen interactions
These were performed after a modified protocol from Zamolodchikov et al.30 Briefly, fibrinogen (Calbiochem; 0.5 mg/mL in 50 mM Tris, 100 mM NaCl at pH 7.4) was incubated with 50 μg Affimer protein F5 before the addition of 35 nM plasmin (Enzyme Research Laboratories, Swansea, United Kingdom) for 1 hour at 37°C to generate fibrinogen degradation products. Digestions were stopped with 40 nM aprotinin (Sigma, Gillingham, United Kingdom). F5-fibrinogen degradation products were pulled down with His-tag isolating beads (Dynabeads; Thermo Fisher Scientific) and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and mass spectrometry to identify F5-binding fibrinogen degradation products (full details in supplemental Methods).
Molecular modeling
A homology model of Affimer F5 was created using I-TASSER and the Maestro graphical user interface to check the validity of the model produced.31 The published crystal structure of the Affimer scaffold (PDB ID: 4N6T) was used as a template to create a model of Affimer F5.19 Docking of this F5 model to the published structure of fibrinogen fragment D (PDB ID: 1FZA)32 was carried out using AutoDock 4.2.33 A total of 100 docking iterations were calculated for each predicted site, using a Lamarckian Genetic Algorithm. The resulting poses were clustered, based on a 2 Å root mean squared deviation. The cluster with the lowest energy conformation, and also the most populated cluster pose, were further examined using PyMOL.34,35
Plasmin generation assay
The rate of plasmin generation by tPA was studied as previously described.29 Additional control assays were performed to investigate effects of Affimer protein on plasmin or tPA activity (details in supplemental Methods).
Platelet function assays
Aggregation in whole blood
Blood was collected in Hirudin Tubes (Roche Diagnostics International Ltd, Rotkreuz, Switzerland), and whole-blood aggregation was performed using the Multiplate analyzer (Dynabyte Medical, Munich, Germany). Briefly, whole blood (300 µL) was diluted 1:2 (vol/vol) with saline solution (0.9% NaCl) containing Affimer protein (Affimer:fibrinogen molar ratio of 10:1) and incubated for 3 minutes at 37°C. Agonists adenosine diphosphate (ADP) or collagen (ADPtest; COLtest, Roche Diagnostics International Ltd; final concentrations of 6.5 µM and 3.2 µg/mL, respectively) were then added. The area under the aggregation curve and maximum aggregation were calculated.
Platelet activation assay
Platelet activation was measured by whole-blood flow cytometry, as previously described.36 Whole-blood samples were incubated with buffer only (phosphate-buffered saline), scaffold, or F5 (Affimer:fibrinogen molar ratio of 10:1), stimulated with 5 µM thrombin receptor activating peptide and stained with platelet marker CD42b-APC and activation marker CD62P-PE (BD Biosciences) for 20 minutes. Samples were run on a Beckman Coulter CytoFLEX RUO Flow Cytometer and analyzed on FlowJo (v10). Automatic compensation was performed with BD CompBeads (BD Biosciences).
Statistical analysis
Statistical analysis was performed using Prism 7 (GraphPad). A P value of less than .05 was considered significant. For turbidimetric, plasmin generation, and permeation experiments, statistical significance between different groups (different molar ratios) and the control was evaluated using 1-way ANOVA and Dunnett’s multiple comparisons test. The statistical significance between Affimers and Affimer scaffold in the different plasma samples of healthy subjects was analyzed using a 2-tailed, paired Student t test. Unpaired Student t test was used to compare buffer-only controls in normal and FVIII-deficient plasma. Unless otherwise stated, the data are presented as mean ± SD.
Results
Isolation of fibrinogen-binding Affimer proteins
The Affimer phage library was screened against fibrinogen to identify fibrinogen-binding Affimer reagents. After 3 rounds of panning, a total of 80 different clones were picked from 2 screens; of these, 15 were found to have distinct sequences and were subcloned into pET11a for large-scale production.
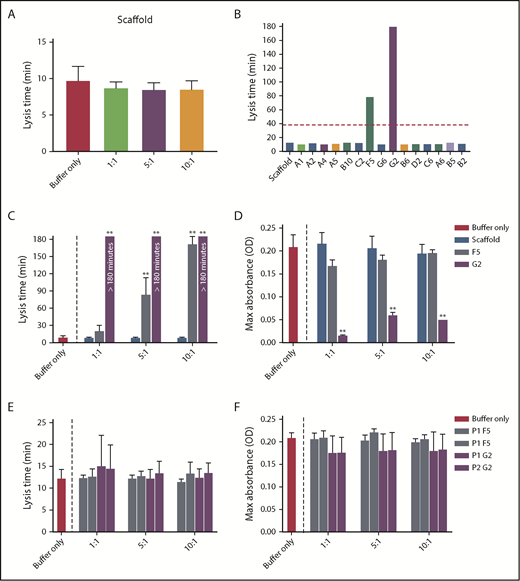
Increasing concentrations of Affimer scaffold showed no effect on plasma clot lysis and was used as a negative control in all subsequent experiments (Figure 1A). All 15 fibrinogen-binding Affimer proteins were then tested initially, using an Affimer:fibrinogen molar concentration of 5:1, with Affimers F5 and G2 showing more than 3-fold prolongation in plasma clot lysis (Figure 1B).
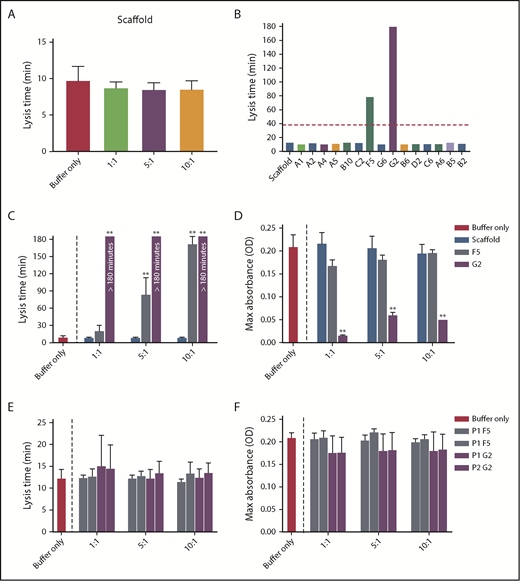
Selection of fibrinogen-binding Affimers after phage panning. After phage panning, fibrinogen-binding Affimers were tested in turbidimetric assays in a plasma system to determine effects of the Affimers on clot formation and lysis. (A) The effect of scaffold-only control protein and (B) 15 Affimer proteins with distinct sequences on fibrin clot lysis (B). The red dotted line shows threefold increase in prolongation of clot lysis. (C) The effect of F5 and G2 on fibrin clot lysis and (D) clot maximum absorbance in human plasma turbidimetric assays. (E) The effect of linear peptides of the 2 variable regions of Affimer proteins F5 (P1F5 and P2F5) and G2 (P1G2 and P2G2) on fibrin clot lysis and (F) clot maximum absorbance. Numbers on the x-axis (A,C-F) represent Affimer protein:fibrinogen molar ratios. Data are presented as the mean ± SD of 3 independent experiments. Statistical analysis was performed using 1-way ANOVA, comparisons made against buffer-only control (A,C-F) or scaffold (B). **P< .01.
Selection of fibrinogen-binding Affimers after phage panning. After phage panning, fibrinogen-binding Affimers were tested in turbidimetric assays in a plasma system to determine effects of the Affimers on clot formation and lysis. (A) The effect of scaffold-only control protein and (B) 15 Affimer proteins with distinct sequences on fibrin clot lysis (B). The red dotted line shows threefold increase in prolongation of clot lysis. (C) The effect of F5 and G2 on fibrin clot lysis and (D) clot maximum absorbance in human plasma turbidimetric assays. (E) The effect of linear peptides of the 2 variable regions of Affimer proteins F5 (P1F5 and P2F5) and G2 (P1G2 and P2G2) on fibrin clot lysis and (F) clot maximum absorbance. Numbers on the x-axis (A,C-F) represent Affimer protein:fibrinogen molar ratios. Data are presented as the mean ± SD of 3 independent experiments. Statistical analysis was performed using 1-way ANOVA, comparisons made against buffer-only control (A,C-F) or scaffold (B). **P< .01.
F5 significantly increased clot lysis time in a concentration-dependent manner, from 9.8 ± 1.8 minutes in the buffer-only control to 84.3 ± 28.8 minutes at 5:1 Affimer:fibrinogen molar ratio and 172.6 ± 12.7 minutes at 10:1 (P < .0001). G2 prevented clot lysis during the 3-hour observation time at molar ratios of 1:1, 5:1, and 10:1 (Figure 1C). G2 significantly reduced clot maximum absorbance at all concentrations, whereas F5 had no significant effect (Figure 1D).
Conformational interactions are responsible for changes in clot structure/lysis
Interindividual variability in the effects of F5 and G2 on plasma clot lysis
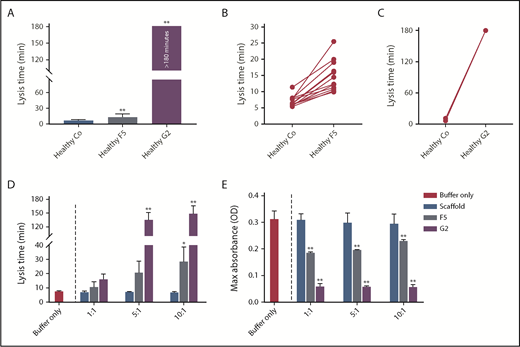
To determine interindividual variability in response, we studied the effects of Affimer F5 and G2 in individual plasma samples from healthy control patients (n = 12). Affimer F5 showed lysis prolongation in all samples (Figure 2A-B), whereas G2 completely prevented clot lysis during the observation period of 3 hours in all individuals (Figure 2A,C).
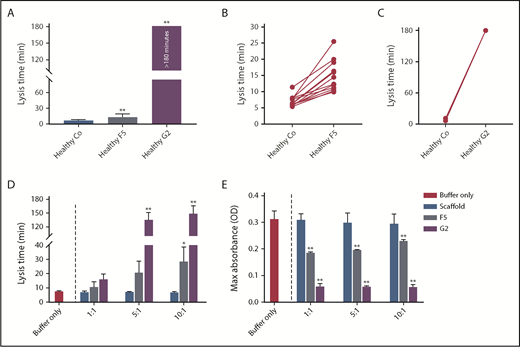
The effect of F5 and G2 on fibrin clot lysis in healthy individuals and in a purified system. (A) Fibrinogen-binding Affimers F5 and G2 were tested in turbidimetric assays in plasma from healthy individuals (n = 12) to study interindividual variability, with scaffold control protein included as a control (Co). Data are presented as the mean ± SD of 12 subjects in each group. (B) Effect of Affimer F5 and (C) Affimer G2 compared with scaffold control protein in the 12 individual plasma samples. The statistical significance of the effect of Affimer F5 or G2 was determined using a 2-tailed, paired Student t test. **Difference from scaffold-only control; P < .01. (D) The effect of F5 and G2 on clot lysis and (E) clot maximum absorbance was tested in turbidimetric assays in a purified fibrinogen system to determine whether effects seen in plasma were fibrinogen-specific. Affimer scaffold-only protein was included as a control. Numbers on the x-axis represent Affimer protein:fibrinogen molar ratios. Data are presented as the mean ± SD of 3 independent experiments. Statistical analysis was performed using 1-way ANOVA. Asterisks represent difference from buffer-only control; *P < .05; **P < .01.
The effect of F5 and G2 on fibrin clot lysis in healthy individuals and in a purified system. (A) Fibrinogen-binding Affimers F5 and G2 were tested in turbidimetric assays in plasma from healthy individuals (n = 12) to study interindividual variability, with scaffold control protein included as a control (Co). Data are presented as the mean ± SD of 12 subjects in each group. (B) Effect of Affimer F5 and (C) Affimer G2 compared with scaffold control protein in the 12 individual plasma samples. The statistical significance of the effect of Affimer F5 or G2 was determined using a 2-tailed, paired Student t test. **Difference from scaffold-only control; P < .01. (D) The effect of F5 and G2 on clot lysis and (E) clot maximum absorbance was tested in turbidimetric assays in a purified fibrinogen system to determine whether effects seen in plasma were fibrinogen-specific. Affimer scaffold-only protein was included as a control. Numbers on the x-axis represent Affimer protein:fibrinogen molar ratios. Data are presented as the mean ± SD of 3 independent experiments. Statistical analysis was performed using 1-way ANOVA. Asterisks represent difference from buffer-only control; *P < .05; **P < .01.
Effects of Affimers on clots made from purified protein
To determine whether the prolongation of lysis was fibrinogen-specific, the Affimer proteins were also tested in turbidimetric assays using purified fibrinogen. Lysis time was increased from 7.6 ± 0.3 minutes in buffer-only control to 28.7 ± 10.1 minutes (P = .0445) and 149.3 ± 16.8 minutes (P < .0001) by F5 and G2, respectively, at an Affimer:fibrinogen molar ratio of 10:1 (Figure 2D). The presence of F5 and G2 decreased the clot maximum absorbance (Figure 2E).
Effect of F5 and G2 on the structure of plasma clots
Confocal and scanning electron microscopy
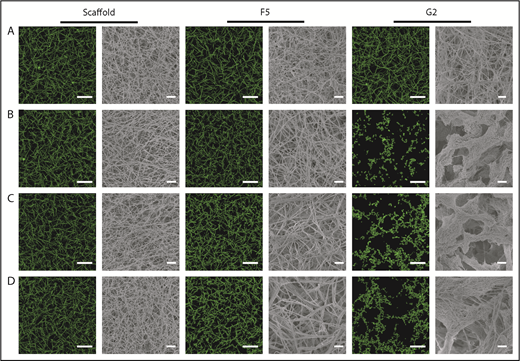
Confocal microscopy was used for visualization of physiological and hydrated clots, whereas clot ultrastructure was analyzed by scanning electron microscopy. Scaffold protein and Affimer F5 maintained physiological clot structure, whereas G2 induced significant changes, resulting in the formation of thinner fibrin fibers gathered into dense tangled bundles (Figure 3), explaining the large reduction in clot maximum absorbance by G2 in turbidimetric assays.
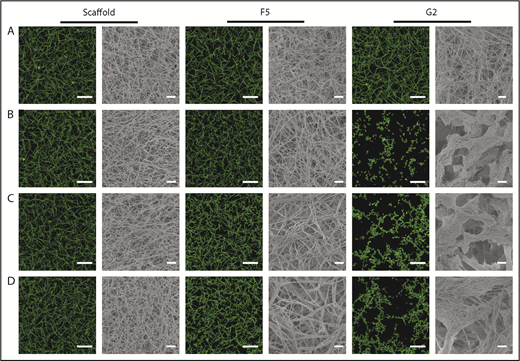
The effect of Affimers F5 and G2 on fibrin network structure. Scaffold and Affimer proteins F5 and G2 were added to plasma clots and visualized using laser scanning confocal microscopy and scanning electron microscopy. For confocal images, clots were made with the addition of fluorescently labeled Alexa Fluor-488 fibrinogen. Z stacks of 30 slices over 20.30 µm were taken, 3-dimensional images of compiled Z stacks are presented. Clots were made with increasing concentrations of Affimer proteins (increasing Affimer:fibrinogen molar ratio). (A) Buffer-only control, (B) 1:1, (C) 5:1, and (D) 10:1 Affimer:fibrinogen molar ratio. Laser scanning confocal microscopy scale bar = 20 μm; scanning electron microscopy scale bar = 1 μm.
The effect of Affimers F5 and G2 on fibrin network structure. Scaffold and Affimer proteins F5 and G2 were added to plasma clots and visualized using laser scanning confocal microscopy and scanning electron microscopy. For confocal images, clots were made with the addition of fluorescently labeled Alexa Fluor-488 fibrinogen. Z stacks of 30 slices over 20.30 µm were taken, 3-dimensional images of compiled Z stacks are presented. Clots were made with increasing concentrations of Affimer proteins (increasing Affimer:fibrinogen molar ratio). (A) Buffer-only control, (B) 1:1, (C) 5:1, and (D) 10:1 Affimer:fibrinogen molar ratio. Laser scanning confocal microscopy scale bar = 20 μm; scanning electron microscopy scale bar = 1 μm.
Clot permeation
Scaffold protein and F5 had no effect on plasma clot permeability, whereas G2 significantly increased permeability (all at an Affimer:fibrinogen molar ratio of 5:1; Table 1).
Affimer protein–fibrinogen interactions
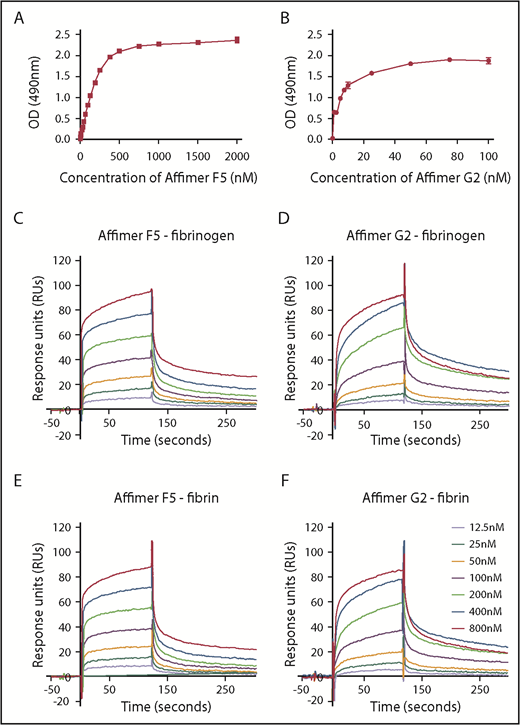
ELISA experiments showed F5 and G2 binding affinity to fibrinogen at KD of 217 ± 47.7 nM and 5.6 ± 0.8 nM, respectively (Figure 4A-B).
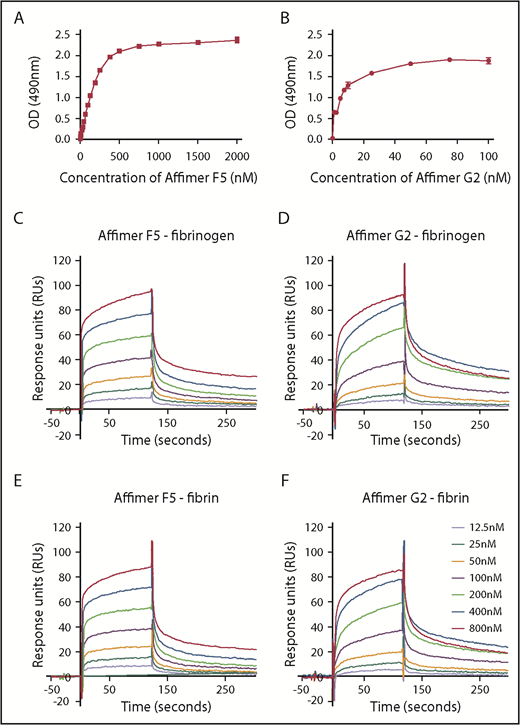
Characterization of Affimer interaction with fibrinogen and fibrin. Binding affinities of Affimers F5 and G2 to fibrinogen were studied using ELISA-based binding assay in which increasing concentrations of the Affimers were added to fibrinogen coated wells before detection of Affimer. (A) Binding of Affimer proteins F5 and (B) G2 to fibrinogen. (C-D) Binding kinetics of Affimer F5 and G2 interaction with fibrinogen and (E-F) fibrin using Biacore SPR. Affimer proteins (12.5-800 nM) were injected over a fibrinogen- or fibrin-derivatized surface before following complex dissociation. Three independent experiments were performed to determine KD values and kinetics. Representative binding data are shown.
Characterization of Affimer interaction with fibrinogen and fibrin. Binding affinities of Affimers F5 and G2 to fibrinogen were studied using ELISA-based binding assay in which increasing concentrations of the Affimers were added to fibrinogen coated wells before detection of Affimer. (A) Binding of Affimer proteins F5 and (B) G2 to fibrinogen. (C-D) Binding kinetics of Affimer F5 and G2 interaction with fibrinogen and (E-F) fibrin using Biacore SPR. Affimer proteins (12.5-800 nM) were injected over a fibrinogen- or fibrin-derivatized surface before following complex dissociation. Three independent experiments were performed to determine KD values and kinetics. Representative binding data are shown.
SPR experiments, using a range of Affimer concentrations, demonstrated that F5 and G2 bound immobilized fibrinogen with KD values of 52 ± 1.3 and 38 ± 6.1 nM, respectively (Figure 4C-D). The Affimers had a similar affinity for fibrin with KD values of 58 ± 9.3 nM and 53 ± 8.2 nM, respectively (Figure 4E-F). Affimer G2 showed slower association rates compared with F5, but dissociation rates were similar (Figure 4C-F). SPR data are summarized in Table 2. The Affimer scaffold or a control Affimer (Affimer directed against SUMO protein22 ) did not bind either fibrinogen or fibrin (supplemental Figure 1).
Identification of potential fibrinogen–Affimer F5 interaction site
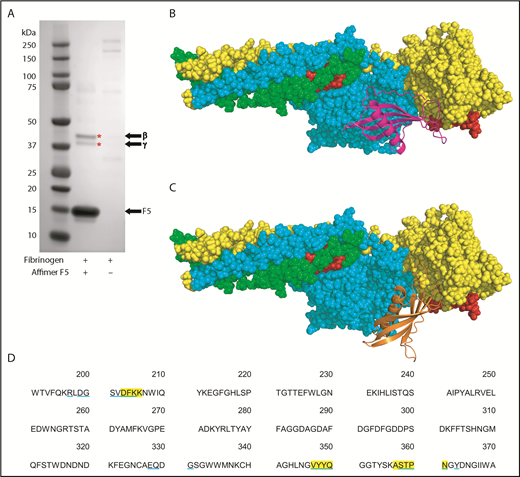
To further characterize the Affimer–fibrinogen interaction, a combination of protein pulldowns and molecular modeling techniques were used. Given that G2-induced nonphysiological changes in clot structure, we concentrated on Affimer F5 for these more in-depth studies. A pulldown assay was performed after incubation of Affimer F5 with fibrinogen and digestion with plasmin to form fibrinogen degradation products. F5 bound to a fibrinogen region, which, when analyzed by SDS-PAGE, contained 2 distinct protein bands at approximately 42 and 38 kDa (Figure 5A), consistent with the molecular weights of the β and γ portion of the fibrinogen D fragment, respectively.37-39 Mass spectrometry confirmed the bands to be composed of fibrinogen β and γ chain.
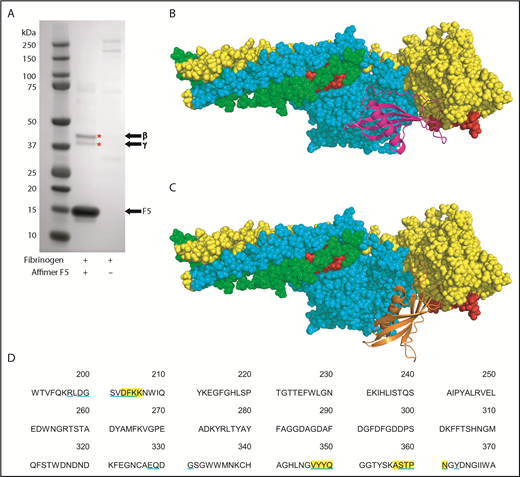
Identification of fibrinogen fragment D as site of F5 interaction. (A) Affimer F5 was incubated with fibrinogen before plasmin digest, and pulldown of F5-fibrinogen degradation products with His-tag isolating beads. Proteins marked with an asterisk (*) were identified by mass spectrometry (LC-MS/MS). The position of Affimer F5 on the gel is marked. Gel image is representative of 3 independent experiments. (B-C) Autodock 4.2 prediction of Affimer F5 binding to the γ chain of the D fragment of fibrinogen. Fibrinogen fragment D is shown as a space-filling model, with the α chain in green, β chain in turquoise, and γ chain in yellow. The tPA/plasminogen binding site α148-160 and the tPA binding site γ312-324 are highlighted in red. (B) In the lowest-energy pose, the variable region loops of F5 (F5 depicted with magenta ribbons) are interacting with the fibrinogen γ chain near to the γ312-324 tPA binding site. (C) The most populated clusters of predicted docking poses also placed Affimer F5 (orange ribbons) to a similar area of fibrinogen fragment D. (D) Amino acid sequence of fibrinogen γ chain, with those amino acids within 4 Å of Affimer F5′s variable region loops marked. Fibrinogen residues close to Affimer loops in the lowest-energy pose are highlighted yellow, and those in the most populated pose are underlined blue.
Identification of fibrinogen fragment D as site of F5 interaction. (A) Affimer F5 was incubated with fibrinogen before plasmin digest, and pulldown of F5-fibrinogen degradation products with His-tag isolating beads. Proteins marked with an asterisk (*) were identified by mass spectrometry (LC-MS/MS). The position of Affimer F5 on the gel is marked. Gel image is representative of 3 independent experiments. (B-C) Autodock 4.2 prediction of Affimer F5 binding to the γ chain of the D fragment of fibrinogen. Fibrinogen fragment D is shown as a space-filling model, with the α chain in green, β chain in turquoise, and γ chain in yellow. The tPA/plasminogen binding site α148-160 and the tPA binding site γ312-324 are highlighted in red. (B) In the lowest-energy pose, the variable region loops of F5 (F5 depicted with magenta ribbons) are interacting with the fibrinogen γ chain near to the γ312-324 tPA binding site. (C) The most populated clusters of predicted docking poses also placed Affimer F5 (orange ribbons) to a similar area of fibrinogen fragment D. (D) Amino acid sequence of fibrinogen γ chain, with those amino acids within 4 Å of Affimer F5′s variable region loops marked. Fibrinogen residues close to Affimer loops in the lowest-energy pose are highlighted yellow, and those in the most populated pose are underlined blue.
Molecular modeling was performed using the crystal structures of fragment D and a homology model of Affimer F5, based on the Affimer scaffold crystal structure.19,32 Using an unbiased docking approach, Autodock 4.2 predicted binding of the Affimer to the γ chain of fibrinogen D fragment in an area spatially close to the γ312-324 tPA and the α148-160 tPA/plasminogen binding sites40-42 (Figure 5B-C). The region bound by F5 was occupied by both the lowest-energy and the most populated clusters of predicted docking poses. The fibrinogen residues within 4 Å (the accepted limit of hydrogen bonding interactions) of the F5 variable region loops were similar in both docking models (Figure 5D).
Affimer protein F5 reduces rate of plasmin generation by a fibrin-dependent mechanism and through interference with tPA–plasminogen interaction
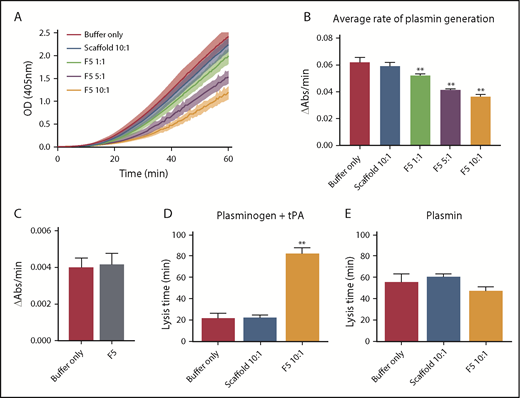
Given the predicted binding of Affimer F5 to fibrinogen in close proximity to both the α148-160 tPA/plasminogen binding site and the γ312-324 tPA binding site, we investigated the effect of F5 on fibrin(ogen)-mediated plasmin generation, using a chromogenic substrate S2251 assay. Affimer F5 significantly reduced the rate of plasmin generation at all molar ratios used from 0.062 ± 0.003 au/min in control samples (buffer only) to 0.052 ± 0.001 (P= .0013), 0.041 ± 0.001 (P < .0001), and 0.036 ± 0.001 (P < .0001) au/min at 1:1, 5:1, 10:1 Affimer:fibrinogen molar ratios, respectively, whereas scaffold control protein had no effect (Figure 6A-B). When no fibrin(ogen) was present, F5 did not alter plasmin generation, confirming the Affimer’s fibrin(ogen) specificity (Figure 6C). The fibrin-specific mode of action of Affimer F5 was further demonstrated by its inability to prolong urokinase-type plasminogen activator-induced lysis (supplemental Figure 2). In a purified turbidimetric assay, F5 prolonged plasminogen-tPA-induced clot lysis (Figure 6D) but not plasmin-induced lysis (Figure 6E), indicating modulation of plasmin generation.
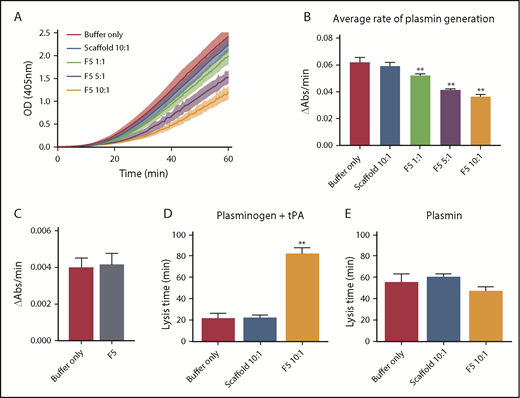
Affimer protein F5 reduced the rate of plasminogen to plasmin conversion. (A-B) The effect of Affimer F5 on plasminogen to plasmin conversion was studied in a plasmin generation assay. Fibrin clots were formed with fibrinogen (0.5 mg/mL) and increasing concentrations of Affimer F5 (Affimer:fibrinogen molar ratios of 1:1, 5:1, and 10:1) with scaffold-only protein included as a control. Clots were lysed by overlaying with tPA in the presence of S2251. (A) Increase in absorbance readings for the first 60 minutes of plasmin generation assays. (B) Plasmin generation presented as rate of chromogenic substrate hydrolysis (au/min). (C) Experiments were also performed without fibrin(ogen), while using the highest concentration of Affimer protein, to assess the fibrin(ogen)-specificity of Affimer F5. (D) In a purified turbidity and lysis assay, preformed clots with and without Affimer protein were overlaid with lysis mix containing either plasminogen–tPA or (E) plasmin. Numbers on the x-axis represent Affimer protein:fibrinogen molar ratios. Data are presented as the mean ± SD of 3 independent experiments. Statistical analysis was performed using 1-way ANOVA, **Difference from buffer-only control; P < .01.
Affimer protein F5 reduced the rate of plasminogen to plasmin conversion. (A-B) The effect of Affimer F5 on plasminogen to plasmin conversion was studied in a plasmin generation assay. Fibrin clots were formed with fibrinogen (0.5 mg/mL) and increasing concentrations of Affimer F5 (Affimer:fibrinogen molar ratios of 1:1, 5:1, and 10:1) with scaffold-only protein included as a control. Clots were lysed by overlaying with tPA in the presence of S2251. (A) Increase in absorbance readings for the first 60 minutes of plasmin generation assays. (B) Plasmin generation presented as rate of chromogenic substrate hydrolysis (au/min). (C) Experiments were also performed without fibrin(ogen), while using the highest concentration of Affimer protein, to assess the fibrin(ogen)-specificity of Affimer F5. (D) In a purified turbidity and lysis assay, preformed clots with and without Affimer protein were overlaid with lysis mix containing either plasminogen–tPA or (E) plasmin. Numbers on the x-axis represent Affimer protein:fibrinogen molar ratios. Data are presented as the mean ± SD of 3 independent experiments. Statistical analysis was performed using 1-way ANOVA, **Difference from buffer-only control; P < .01.
Given the above data, competitive binding assays were performed to determine whether F5 prevented tPA or plasminogen interaction with fibrin. The presence of multiple tPA and plasminogen binding sites within fibrin(ogen) made competition assays using whole fibrin(ogen) difficult to interpret. Therefore, given the results of our pulldown assays and molecular modeling data, all competition assays were undertaken using fibrin DD fragment. F5, tPA, and plasminogen all demonstrated binding to fibrin DD fragment, whereas scaffold protein showed no binding (supplemental Figure 3A-D). F5 had no effect on tPA or plasminogen binding to DD fragment (supplemental Figure 3E-F). Additional control experiments confirmed there was no direct effect of the Affimer on tPA or plasmin protein activity (supplemental Figure 4A-B), and that F5 showed no direct binding to either tPA or plasminogen (supplemental Figure 4C-D).
Clinical relevance
Effect of Affimer protein F5 on fibrinolysis in plasma deficient in FVIII (hemophilia A)
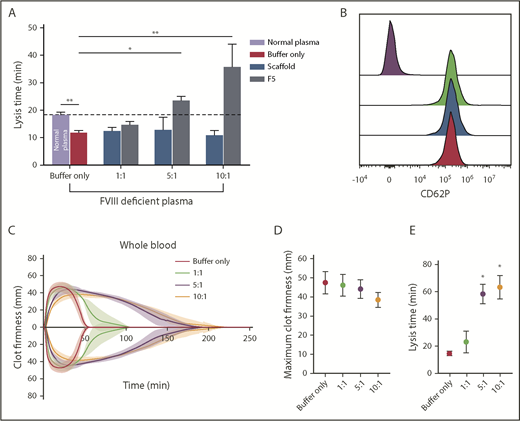
The effects of F5 on fibrinolysis were investigated in FVIII-deficient plasma (consistent with the clinical presentation of hemophilia A) in turbidimetric experiments after initiation of clotting with tissue factor to account for the effects of FVIII deficiency. Similar experiments were performed in normal pooled plasma (control clots, supplemental Figure 5A). In control clots, lysis time in buffer-only control was 18.37 ± 0.9 min, and was significantly shorter in FVIII-deficient plasma at 11.83 ± 0.8 minutes (P= .0009; Figure 7A). The addition of Affimer F5 to FVIII deficient plasma prolonged lysis from 11.83 ± 0.8 to 23.77 ± 1.2 minutes (P = .0105) and 36.77 ± 8.2 minutes (P < .0001) at 5:1 and 10:1 Affimer:fibrinogen molar ratios, thus normalizing, and even prolonging, the short lysis time in this condition (Figure 7A). Clotting time in FVIII-deficient plasma (time from start of the reaction until maximum absorbance is reached) was not altered by Affimer F5 (supplemental Figure 6B). Prolongation of lysis by F5 was consistent when different concentrations of tissue factor (0.5-10 pM) were used to initiate thrombin generation and clot formation in FVIII-deficient plasma (supplemental Figure 5C).
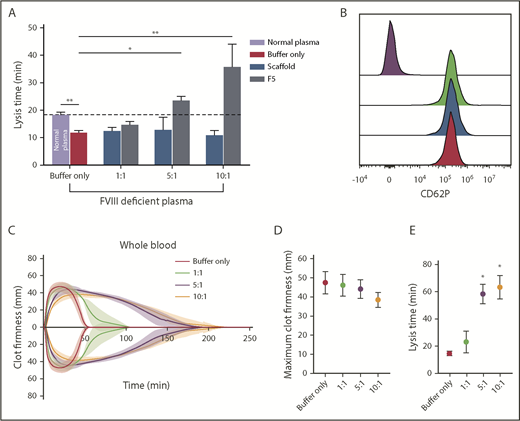
The effect of Affimer F5 on clot lysis in FVIII-deficient plasma, and on lysis and platelet activation in whole blood. (A) Turbidimetric experiments in which clot formation was initiated with tissue factor (5 pM) were performed using normal plasma and FVIII-deficient plasma. The effect of increasing concentrations of Affimer F5 were tested in FVIII-deficient plasma, with scaffold-only protein included as a control. Numbers on the x-axis represent Affimer:fibrinogen molar ratios. Data are presented as the mean ± SD of 3 independent experiments. Unpaired Student t test was used to compare buffer-only controls in normal and FVIII-deficient plasma; **P < .01. 1-way ANOVA was used to determine the significance of Affimer F5 effect in FVIII-deficient plasma at different concentrations. Asterisks represents difference from buffer-only control; *P < .05; **P < .01. (B) The effect of F5 on platelet activation was tested using whole blood from 3 healthy volunteers. Blood was incubated with buffer only (green peak), scaffold (blue peak), or F5 (red peak) and then stimulated with thrombin receptor activating peptide. CD62P expression was compared with basal platelets (purple peak). Affimer proteins were used at 10:1 Affimer:fibrinogen molar ratio. (C) The effect of increasing concentrations of Affimer F5 on whole-blood lysis was tested using human whole blood from 3 healthy individuals using ROTEM. Tissue factor and tPA were used for initiation of clotting and lysis, respectively. (D) Maximum clot firmness from ROTEM experiments. (E) Lysis time of whole-blood clots formed in the presence of Affimer F5. Numbers on the x-axis represent Affimer protein:fibrinogen molar ratios. Data are presented as the mean ± SD of 3 independent experiments. Statistical analysis was performed using 1-way ANOVA. Asterisks represents difference from buffer-only control; *P < .05; **P < .01.
The effect of Affimer F5 on clot lysis in FVIII-deficient plasma, and on lysis and platelet activation in whole blood. (A) Turbidimetric experiments in which clot formation was initiated with tissue factor (5 pM) were performed using normal plasma and FVIII-deficient plasma. The effect of increasing concentrations of Affimer F5 were tested in FVIII-deficient plasma, with scaffold-only protein included as a control. Numbers on the x-axis represent Affimer:fibrinogen molar ratios. Data are presented as the mean ± SD of 3 independent experiments. Unpaired Student t test was used to compare buffer-only controls in normal and FVIII-deficient plasma; **P < .01. 1-way ANOVA was used to determine the significance of Affimer F5 effect in FVIII-deficient plasma at different concentrations. Asterisks represents difference from buffer-only control; *P < .05; **P < .01. (B) The effect of F5 on platelet activation was tested using whole blood from 3 healthy volunteers. Blood was incubated with buffer only (green peak), scaffold (blue peak), or F5 (red peak) and then stimulated with thrombin receptor activating peptide. CD62P expression was compared with basal platelets (purple peak). Affimer proteins were used at 10:1 Affimer:fibrinogen molar ratio. (C) The effect of increasing concentrations of Affimer F5 on whole-blood lysis was tested using human whole blood from 3 healthy individuals using ROTEM. Tissue factor and tPA were used for initiation of clotting and lysis, respectively. (D) Maximum clot firmness from ROTEM experiments. (E) Lysis time of whole-blood clots formed in the presence of Affimer F5. Numbers on the x-axis represent Affimer protein:fibrinogen molar ratios. Data are presented as the mean ± SD of 3 independent experiments. Statistical analysis was performed using 1-way ANOVA. Asterisks represents difference from buffer-only control; *P < .05; **P < .01.
Effect of Affimer protein F5 on platelet function and fibrinolysis in whole blood
To further investigate specificity, we tested the effects of Affimer F5 on whole-blood aggregation, and no effects were observed on ADP- or collagen-induced platelet aggregation (Table 3). In addition, platelet CD62P expression was not affected by either Affimer F5 or scaffold in resting (data not shown) or thrombin receptor activating peptide-stimulated platelets (Figure 7B).
F5 was tested in whole-blood samples from healthy control patients to ensure that cellular elements did not compromise the antifibrinolytic properties of the Affimer protein. Affimer F5 caused a significant concentration-dependent prolongation of lysis time without significantly affecting maximum clot firmness (Figure 7C-D). Lysis time was prolonged from 14.6 ± 1.6 minutes in the absence of Affimer to 58.3 ± 7.0 minutes (P = .0168) and 63.3 ± 8.7 minutes (P = .0264) at 5:1 and 10:1 Affimer:fibrinogen molar ratio, respectively (Figure 7E), suggesting that molar ratios above 5:1 have few additional effects.
Discussion
Our proof-of-concept study shows that a novel methodology involving small proteins has the ability to prolong clot lysis and stabilize the fibrin network with the potential for clinical development. This offers the opportunity to treat bleeding typically seen after accidental or surgical trauma, as well as hematological abnormalities affecting clotting factors.
Of the 15 distinct fibrinogen-binding Affimer proteins identified, F5 and G2 showed a significant effect on clot lysis and were both found to participate in high-affinity interactions with fibrinogen and fibrin. G2 showed largely similar KD values for fibrinogen binding in both ELISA and SPR, but some minor discrepancies were detected with Affimer F5 that may reflect the different adsorption on a plastic surface for ELISA vs derivatization of the SPR dextran layer.
Although the effects of G2 on lysis were consistent, it caused nonphysiological changes to clot structure, limiting its suitability for development for clinical use. In contrast, Affimer F5 had a consistent antifibrinolytic effect while maintaining a more physiological clot structure. Moreover, there was limited interindividual variability in response to F5 suggesting the Affimer behaves similarly across different individuals. Importantly, Affimer F5 successfully reversed the pathological enhancement of clot lysis in plasma deficient in FVIII, representing individuals with hemophilia A, indicating it has the capability to work in pathological conditions. This would be particularly helpful in more challenging cases in which patients with hemophilia develop antibodies against FVIII, which limit the usefulness of coagulation protein replacement.43,44 The effects of Affimer F5 were maintained in whole blood, indicating that cellular elements do not impair the antifibrinolytic properties of this Affimer. The increase in clot lysis time by F5 ranged from 2- to 10-fold, depending on the concentration of the Affimer used. Recent studies in bleeding and thrombotic disorders indicate that less than 50% difference in clot lysis time can be clinically significant,45-48 and therefore the effects of Affimer F5 are meaningful for future clinical use.
Given the preservation of clot structure by Affimer F5 and the consistent effects on clot lysis, we probed into the mechanisms of this Affimer. We demonstrated that Affimer F5 binds to fragment D of fibrinogen, and molecular modeling simulations predicted binding to fibrinogen γ-chain, in close proximity to the γ312-324 tPA binding site as well as the α148-160 tPA/plasminogen binding site. Although the latter site binds tPA and plasminogen with similar affinity (∼KD 1µM),49 it would be saturated with plasminogen under physiological conditions resulting from the molar excess of this molecule in the circulation.50 Our data indicate that F5 does not alter binding of tPA or plasminogen to fibrin(ogen), at least to DD fragment. Given that the effects of F5 are fibrin-dependent, the most likely explanation is that Affimer F5 interferes with tPA–plasminogen interaction once these proteins are bound to the fibrin clot. This concept is supported by our molecular modeling studies suggesting that F5 binds fibrin(ogen) in an area spatially situated between tPA and plasminogen binding sites in the D region. Further definitive work would be required to confirm this mode of action of F5, which is perhaps best conducted using crystallography studies of fibrin(ogen) in complex with the Affimer and tPA/plasminogen. The fibrin-dependent mode of action of Affimer F5 was confirmed by the absence of an effect when uPA was used to initiate clot lysis.51 This is a particular strength, as any unwanted thrombotic event with clinical use of F5 could be managed with fibrinolytic agents that convert plasminogen to plasmin independent of fibrin, such as urokinase and streptokinase. A constant difficulty in developing anticoagulant or clot-stabilizing agents is the absence of an “antidote” in the event of bleeding or thrombotic complications, respectively. Given the mode of action of F5, we already have a reversing agent, should unwanted thrombosis occur during future in vivo use.
A strength of this study is the development of a novel methodology to stabilize the fibrin clot that may have future clinical implications. The advantage of this technique over human-derived fibrin sealants is the low cost associated with production of Affimer proteins, the low risk for infection, the “single agent” approach, and the fibrin specificity, which minimizes the risks for uncontrolled intravascular coagulation. From the clinical point of view, Affimer proteins may be used directly, or alternatively, may provide a tool for identification of new therapeutic targets that can be subsequently modulated using small molecules.
However, there are limitations to this study, including that only a limited number of fibrinogen-binding Affimer proteins were picked, and therefore larger screens may be required, including investigation of Affimer proteins from earlier panning rounds and lower affinities. This will provide a larger number of Affimer proteins that may be more suitable for clinical use. Second, although the ex vivo studies show a consistent effect for F5, which is promising, in vivo work is yet to be conducted using animal models of bleeding. This work will be a necessary next step to investigate the in vivo safety and efficacy of Affimers.
In conclusion, we present in this proof of concept study a novel methodology to alter fibrinolysis and stabilize the fibrin clot that may offer a simple and affordable way to limit bleeding after traumatic vessel injury or in pathological conditions with inherent abnormalities in the fibrin networks.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank M. Deery and the Cambridge Centre for Proteomics (Department of Biochemistry, University of Cambridge) for identification of proteins with mass spectrometry.
The authors also thank Avacta Life Sciences Ltd., the British Heart Foundation, and Diabetes UK for supporting this work. Biacore experiments were performed in the Wellcome Trust-funded Biomolecular Interactions facility, University of Leeds.
Authorship
Contribution: K.J.K. isolated Affimer F5, performed all F5 experiments and data analysis unless otherwise stated, and performed confocal microscopy work; N.P. performed experiments with Affimer G2 and associated data analysis unless otherwise stated, performed SPR experiments, and analyzed data; R.K. and C.T. performed initial screening and isolated Affimer G2; F.P. contributed to turbidimetric experiments and analysis of data; R.C. performed permeation experiments and analysis of data; F.L.M. performed scanning electron microscopy; K.J.S. performed molecular modeling; I.W.M. contributed to SPR data analysis. B.E.J.S. performed flow cytometry platelet studies; F.L.M., K.J.S., I.W.M., K.A.S., B.E.J.S., K.M.N., R.A.S.A., M.J.M., and D.C.T. contributed to discussion and interpretation of results; K.J.K., N.P., and R.A.A. planned the experiments, interpreted the data, and wrote the manuscript; R.A.A. conceived the project and obtained funding; and all authors critically reviewed the manuscript.
Conflict-of-interest disclosure: M.J.M. and D.C.T. are co-inventors of the Adhiron/Affimer technology and own personal shares in Avacta Life Sciences. The Adhiron/Affimer patent (patent application number PCT/GB2014/050435) is owned by the University of Leeds and licensed to Avacta Ltd. N.P. and R.A.A. received research funding from Avacta Life Sciences. The remaining authors declare no competing financial interests.
Correspondence: Nikoletta Pechlivani, Leeds Institute for Cardiovascular and Metabolic Medicine, LIGHT Laboratories, Clarendon Way, University of Leeds, Leeds LS2 9JT, United Kingdom; e-mail: n.pechlivani@leeds.ac.uk.