In this issue of Blood, provide compelling evidence in mouse models for the roles of the nuclear vitamin D receptor (VDR) and bone marrow macrophages as critical downstream effectors of bone marrow fibrosis.1
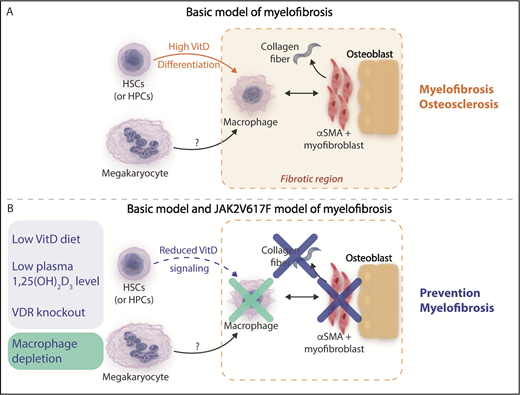
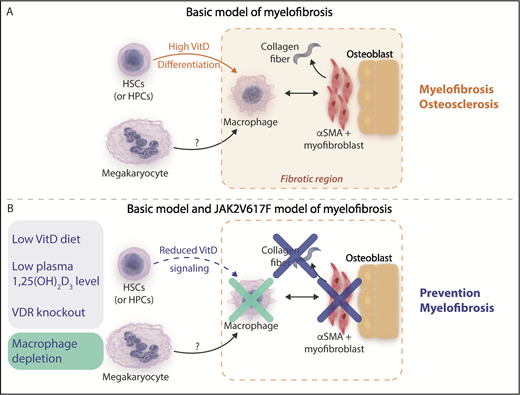
(A) Basic model of myelofibrosis. Increased vitamin D (VitD) signaling resulting from high circulating 1,25(OH)2 vitamin D3 levels acting on transplanted VDR+/+ hematopoietic stem cells (HSCs) and hematopoietic progenitor cells (HPCs) facilitates bone marrow macrophage differentiation. Bone marrow macrophages are proposed to be critical mediators of the development of bone marrow fibrosis acting in concert with other cells observed in fibrotic areas such as osterix+ α-smooth muscle actin–positive (osterix+αSMA+) myofibroblasts and osteoblasts and potentially downstream of megakaryocytes, which were not observed in regions of fibrosis. (B) Mitigation of myelofibrosis in the basic and JAK2V617F transplantation models of myelofibrosis. Reducing VitD signaling by reducing circulating 1,25(OH)2 vitamin D3 levels using a diet low in vitamin D or genetic deletion of the VitD receptor (blue panel) or direct depletion of macrophages (green panel) prevents the development of myelofibrosis in the basic model and a JAK2V617F transplantation model of myelofibrosis. Professional illustration by Somersault18:24. This figure is based on Figure 5C in the article by Wakahashi that begins on page 1619.
(A) Basic model of myelofibrosis. Increased vitamin D (VitD) signaling resulting from high circulating 1,25(OH)2 vitamin D3 levels acting on transplanted VDR+/+ hematopoietic stem cells (HSCs) and hematopoietic progenitor cells (HPCs) facilitates bone marrow macrophage differentiation. Bone marrow macrophages are proposed to be critical mediators of the development of bone marrow fibrosis acting in concert with other cells observed in fibrotic areas such as osterix+ α-smooth muscle actin–positive (osterix+αSMA+) myofibroblasts and osteoblasts and potentially downstream of megakaryocytes, which were not observed in regions of fibrosis. (B) Mitigation of myelofibrosis in the basic and JAK2V617F transplantation models of myelofibrosis. Reducing VitD signaling by reducing circulating 1,25(OH)2 vitamin D3 levels using a diet low in vitamin D or genetic deletion of the VitD receptor (blue panel) or direct depletion of macrophages (green panel) prevents the development of myelofibrosis in the basic model and a JAK2V617F transplantation model of myelofibrosis. Professional illustration by Somersault18:24. This figure is based on Figure 5C in the article by Wakahashi that begins on page 1619.
Primary or secondary myelofibrosis is a chronic myeloproliferative disease driven by JAK2V617F, MPL, or CALR mutations in the majority of patients. Excessive MPL and JAK2 signaling arising from these mutations and the resultant megakaryocytosis observed in this disease are believed to underlie the development of bone marrow fibrosis.2 However, questions remain regarding how similar driver mutations result in the spectrum of diseases that are observed in Philadelphia chromosome–negative myeloproliferative diseases, and the mechanisms by which myelofibrosis develops.
Early histologic studies identified marked increases in bone marrow macrophages in primary myelofibrosis compared with other chronic myeloproliferative diseases. This observation led to speculation that macrophages may play a role in the development of fibrotic disease in humans.3 However, the evidence for such a role remained controversial, and potential drivers of macrophage expansion were unknown.
Wakahashi et al investigated a mouse model of myelofibrosis and osteosclerosis referred to as the “basic model” (see figure panel A) generated when wild-type vitamin D receptor (VDR+/+) donor marrow is transplanted into VDR−/− recipients. This model results in high circulating 1,25(OH)2 vitamin D3 levels that act on transplanted donor VDR+/+ hematopoietic cells. Careful characterization of the fibrotic component within the bone marrow demonstrated persistence of VDR+/+ donor-derived F4/80+ macrophages despite progressive loss of donor stem and progenitor cell potential. Along with VDR+/+ donor-derived macrophages, recipient osterix+ α-smooth muscle actin–positive (osterix+αSMA+) myofibroblasts and osteoblasts were also identified in fibrotic bone marrow areas. This led the authors to hypothesize that generation of collagen fibrosis in this model may be dependent on these cell types.
By using the basic model and an independent transgenic model of JAK2V617F-driven primary myelofibrosis, an elegant series of experiments next demonstrated that VDR signaling and macrophages directly contributed to bone marrow fibrosis. Cohorts of mice fed diets low in vitamin D or in which VDR signaling was genetically abrogated by deletion of the VDR demonstrated mitigation of myelofibrosis in both models, and splenomegaly in the transgenic JAK2V617F model was also moderated. Prevention of marrow fibrosis was also observed in both mouse models when macrophages were depleted (see figure panel B). Importantly, the reduction in myelofibrosis occurred while megakaryocyte numbers in both models were unaffected. These findings suggest that macrophages act as critical downstream effectors of bone marrow fibrosis and that VDR signaling is an important novel mediator in this process.
The authors propose a model in which signaling via the VDR receptor in cKit+ hematopoietic stem and progenitor cells drives macrophage differentiation, whereupon bone marrow macrophages serve as critical mediators of myelofibrosis by acting in concert with stromal cells, such as osterix+αSMA+ myofibroblasts and osteoblasts, and potentially downstream of megakaryocytes. This model is in keeping with a previous study demonstrating that neoplastic monocyte–derived fibrocytes are important mediators of human myelofibrosis.4
Contrary to the predictions of the basic murine model, secondary myelofibrosis in the setting of severe vitamin D deficiency (rickets) that is reversible with vitamin D supplementation in the pediatric setting has been described.5 These cases may well be driven by increased levels of parathyroid hormone that is also significantly increased in VDR−/− mice. In addition, in observational studies of human patients with primary myelofibrosis, vitamin D insufficiency does not seem to influence overall survival.6 Despite these caveats, the study by Wakahashi et al provides significant novel insights into the pathogenesis of myelofibrosis and a rationale for pursing novel treatment strategies in human disease.
Why bone marrow macrophages and neoplastic monocyte–derived fibrocytes are observed to be increased in primary myelofibrosis compared with other chronic myeloproliferative diseases and whether this phenomenon is directly related to VDR signaling remain to be definitively determined. The proposal that VDR signaling in malignant hematopoietic stem and progenitor cell populations may be a driver of macrophage differentiation and fibrotic disease is supported by prior evidence that the acquired driver mutation can be identified in neoplastic macrophage–derived fibrocytes.4 We may therefore hypothesize that there may be a unique role for vitamin D signaling in the neoplastic clone, which is responsible for the development of fibrosis. It would be intriguing to investigate polymorphisms affecting vitamin D biology7 in the development of myelofibrosis in patients with JAK2V617F and other mutations such as those in MPL- and CALR-driven disease. It should also be noted that although the authors argue against VDR being a direct nuclear signal in lineage-positive bone marrow macrophages, contrary evidence exists with regard to VDR expression in this population.8
Overall, Wakahashi et al provide important insights into the pathogenesis of bone marrow fibrosis, including in a JAK2V617F-driven model that has significant implications for human disease. Future research into the biology of primary myelofibrosis and identification of novel targets and therapeutic strategies to prevent bone marrow failure in patients with early-stage myelofibrosis will be stimulated by these findings. Compelling avenues for future translational and clinical research include identifying and targeting molecular pathways of VDR signaling that drive the myelofibrosis phenotype, investigating potential interactions between megakaryocytes and macrophages in primary and secondary myelofibrosis, and targeting macrophages and potentially osterix+αSMA+ myofibroblasts, monocyte-derived fibrocytes, and osteoblasts to prevent development of myelofibrosis.
How best to investigate novel strategies for late-stage fibrotic disease remains an important question. In preclinical models and case reports, bisphosphonates,9 which deplete bone marrow macrophages, may mitigate development of myelofibrosis. Single-agent zometa, however, did not demonstrate consistent clinical efficacy in established primary or secondary myelofibrosis.10 Better stratification and identification of patients who may respond to novel therapies and combinations of different strategies may be needed.
It is also worth noting that although mice fed a diet low in vitamin D in the basic model demonstrated mitigation of fibrosis and correction of hematologic parameters, a significant number of deaths was observed in this cohort which should serve as a cautionary signal when considering potential therapeutic strategies for future clinical trials.
Conflict-of-interest disclosure: A.P.N. declares no competing financial interests.