

In this issue of Blood, report that 21 (29%) of 73 patients with congenital thrombotic thrombocytopenic purpura (TTP) had persistent symptoms of headache, lethargy, and abdominal pain, without thrombocytopenia, that responded to prophylactic plasma.1
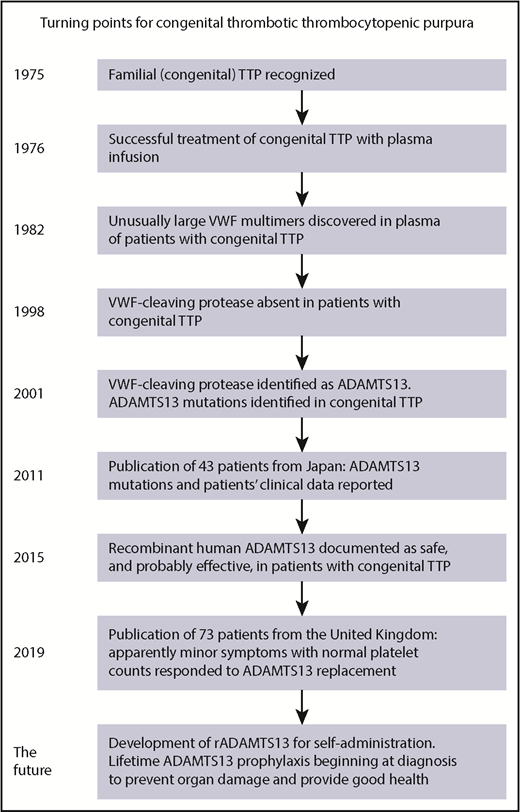
Important turning points in the history of congenital TTP are illustrated. The most important turning point for patients with congenital TTP may be in the near future. Documentation of silent organ injury together with the development of recombinant ADAMTS13 could provide the evidence and the method for lifetime ADAMTS13 replacement beginning at the time of diagnosis. VWF, von Willebrand factor.
Important turning points in the history of congenital TTP are illustrated. The most important turning point for patients with congenital TTP may be in the near future. Documentation of silent organ injury together with the development of recombinant ADAMTS13 could provide the evidence and the method for lifetime ADAMTS13 replacement beginning at the time of diagnosis. VWF, von Willebrand factor.
Fifty years passed after TTP was initially described in 1924 before familial TTP was recognized (see figure).2 In 1976, the effectiveness of plasma infusion was reported; before this report, patients with congenital TTP died.3 In 1982, unusually large multimers of von Willebrand factor were discovered in the plasma of 4 patients with congenital TTP. In 2001, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) was identified and ADAMTS13 mutations were documented in patients with congenital TTP. In 2011, Fujimura et al4 reported ADAMTS13 mutations in 43 patients together with descriptions of their clinical features. This report identified 2 periods of critical risk during the lives of patients with congenital TTP: the first hours after birth and during pregnancy. In 18 patients (42%), the initial critical episode was severe hemolysis in the first hours after birth. Nine patients (21%) were diagnosed when they had acute episodes during pregnancy.
With this background, Alwan and colleagues report their experience with 73 patients diagnosed with congenital TTP in the United Kingdom during the past 15 years. Clinical features varied from mild to fatal. Headaches, often with migraine symptoms, and lethargy were common. Fourteen patients (19%) had a stroke, and stroke was fatal in 4 patients. Three patients had chronic kidney disease. Forty-nine patients (67%) were managed with prophylactic treatment, either plasma or a factor VIII concentrate containing ADAMTS13. Twenty-four patients began prophylactic plasma for apparently minor symptoms such as headache, lethargy, and abdominal pain, without anemia or thrombocytopenia; in 21 patients (88%), the symptoms resolved and their median platelet count increased from 261 000/µL to 325 000/µL. These data illustrate that in addition to acute episodes of hemolysis and thrombocytopenia, comparable to the acute episodes of acquired TTP, patients with congenital TTP also have chronic disease manifested by apparently minor symptoms.
A concurrent report of 120 patients from the International Hereditary TTP Registry (www.ttpregistry.net) describes similar data.5 Among these patients, 25 (21%) reported a stroke; 5 additional patients (4%) reported transient cerebral ischemic symptoms. The age when strokes occurred is not described in either of these 2 reports.5 Previous reports have described strokes in children as young as age 2 years.4 A patient who I follow had a stroke at age 1 day, before exchange transfusion for severe hemolysis and thrombocytopenia was begun.
The series discussed by Alwan et al and other series4,5 with a total of 236 patients provide an understanding of the pathogenesis of congenital TTP, an overview of the clinical features, and the effectiveness of ADAMTS13 replacement. However, they do not provide the individual patient data required for understanding the risks for serious complications, when they may occur, and the patients’ duration of survival. Understanding risks would provide rationale for when to begin lifetime ADAMTS13 replacement treatment. If a newborn infant has severe hemolysis and thrombocytopenia requiring exchange transfusion, it may be appropriate to begin lifetime replacement treatment at that time. But a newborn infant can have severe hemolysis and thrombocytopenia and then be asymptomatic for many years. Does this mean that she has no risk for silent thrombotic organ injury and no need for ADAMTS13 replacement? There are no data for patients with congenital TTP that describe the subtle but serious disorders of cognitive impairment and depression that are common in patients who have recovered from an acute episode of acquired TTP.6 Mild asymptomatic thrombocytopenia, or even a platelet count within the laboratory’s normal range but lower than the patient’s individual normal range,7 may signal the occurrence of platelet consumption and asymptomatic thrombosis. However, prophylactic plasma given at 1- to 2-week intervals is a substantial burden, requiring clinic visits and often placement of an infusaport catheter, which has risks of thrombosis and infection. For these logistical and safety reasons, prophylactic plasma is often deferred. The risks of deferral are not known.
Recombinant ADAMTS13 (rADAMTS13 developed by Shire/Takeda) has been documented to be safe, with suggestions of effectiveness in a phase 1 study.8 It is anticipated that rADAMTS13 could be self-administered at home. Analogous to the experience with hemophilia prophylaxis, from cryoprecipitate to self-administration of recombinant factor VIII, the transition from clinic administration of plasma to home self-administration of rADAMTS13 may be life-changing.
How soon should ADAMTS13 replacement treatment begin? Should it begin in all patients at the time of their diagnosis to prevent thrombosis and organ injury? There are currently no data to support this practice, but the phase 3 trial of rADAMTS13, beginning in 2019, may provide these data. Patients enrolled in this trial will have brain magnetic resonance imaging to determine whether silent cerebral infarctions (SCIs) have occurred. SCIs can cause cognitive impairment and depression; these disorders can cause the symptoms of fatigue and lethargy described by Alwan and colleagues. SCIs are also a significant risk for stroke.
The possible occurrence of SCIs in patients with congenital TTP is suggested by analogy to sickle cell disease, which is also a life-long disorder characterized by acute episodes of symptomatic thrombosis and continuing subclinical microvascular thrombosis with ischemic organ injury. SCIs are common in sickle cell disease, occurring in 35% of children by age 15 years9 and in at least 50% of adults by age 32 years.10 Identification of SCIs in patients with congenital TTP would provide the rationale for lifetime ADAMTS13 replacement for all patients, beginning at the time of their diagnosis.
Congenital TTP is approaching a critical turning point. Individual patient data should provide evidence for when to begin lifetime ADAMTS13 replacement treatment. Simplicity of home administration of ADAMTS13 replacement may make lifetime treatment acceptable. With appropriate ADAMTS13 replacement, patients with congenital TTP should have long and healthy lives.
Conflict-of-interest disclosure: J.N.G. declares no competing financial interests.

