

Key Points
PEL in individuals with HIV is associated with a Kaposi sarcoma herpesvirus-driven IL-6 and IL-10–related syndrome.
EBV status of the tumor and elevated serum IL-6 levels are prognostic in PEL.

Abstract
Primary effusion lymphoma (PEL) is an aggressive HIV-associated lymphoma with a relatively poor prognosis in the era of effective HIV therapy. Kaposi sarcoma herpesvirus (KSHV) is the etiologic agent, and ∼80% of tumors are coinfected with Epstein-Barr virus (EBV). A better understanding of how KSHV-related immune dysregulation contributes to the natural history of PEL will improve outcomes. Twenty patients with PEL diagnosed between 2000 and 2013, including 19 treated with modified infusional etoposide, vincristine, and doxorubicin with cyclophosphamide and prednisone (EPOCH), were identified. We compared their clinical, virologic, and immunologic features vs 20 patients with HIV-associated diffuse large B-cell lymphoma and 19 patients with symptomatic interleukin (IL)-6 related KSHV-associated multicentric Castleman disease. Survival analyses of treated patients with PEL were then performed to identify prognostic factors and cancer-specific mortality. Compared with HIV-associated diffuse large B-cell lymphoma, PEL was associated with significant hypoalbuminemia (P < .0027), thrombocytopenia (P = .0045), and elevated IL-10 levels (P < .0001). There were no significant differences in these parameters between PEL and KSHV-associated multicentric Castleman disease. Median overall survival in treated patients with PEL was 22 months, with a plateau in survival noted after 2 years. Three-year cancer-specific survival was 47%. EBV-positive tumor status was associated with improved survival (hazard ratio, 0.27; P = .038), and elevated IL-6 level was associated with inferior survival (hazard ratio, 6.1; P = .024). Our analysis shows that IL-6 and IL-10 levels contribute to the natural history of PEL. Inflammatory cytokines and tumor EBV status are the strongest prognostic factors. Pathogenesis-directed first-line regimens are needed to improve overall survival in PEL.
Introduction
Primary effusion lymphoma (PEL) is an aggressive B-cell non-Hodgkin lymphoma with a unique clinical presentation and gene expression profile.1 It was originally described as an effusion lymphoma in patients with HIV; however, extracavitary presentations have subsequently been noted. Kaposi sarcoma herpesvirus (KSHV), also known as human herpesvirus-8, is the etiologic agent of PEL as well as Kaposi sarcoma and a form of multicentric Castleman disease (KSHV-MCD).2-4 Approximately 80% of PEL cases are coinfected with Epstein-Barr virus (EBV) and KSHV. Given this common viral etiology, patients with PEL may have concurrent Kaposi sarcoma and/or KSHV-MCD. There is currently a poor understanding of the disease pathogenesis of this rare lymphoma whose incidence may be increasing in the era of modern antiretroviral therapy (ART).5
KSHV is notable for its ability to cause dysregulation of the host immune system, which may contribute to disease pathogenesis and the unique clinical manifestations of KSHV-associated malignancies. In KSHV-MCD, patients have an elevated circulating KSHV viral load and KSHV-associated B-cell lymphoproliferation leading to adenopathy, splenomegaly, and a range of systemic inflammatory symptoms. Human interleukin-6 (IL-6) and IL-10 and the KSHV-encoded homolog viral IL-6 (vIL-6) are implicated in pathogenesis.6,7 Treatment of KSHV-MCD with the monoclonal anti-CD20 antibody rituximab leads to long-term disease remission in most patients and dramatically improves survival.8-10
Cytokine-related severe systemic inflammation has also been described in HIV/KSHV coinfected patients with no histopathologic characteristics of KSHV-MCD, and this condition has been labeled Kaposi sarcoma herpesvirus inflammatory cytokine syndrome (KICS).11 These patients exhibit elevated IL-6, vIL-6, IL-10, and KSHV viral loads, and a working definition of KICS based on clinical features and KSHV viral load has been proposed (Figure 1). KICS commonly occurs in the setting of suppressed CD4+ T-cell counts but is not attributable to uncontrolled HIV viremia.12 Most patients with KICS have concurrent Kaposi sarcoma and/or PEL, and its diagnosis carries a poor prognosis.

PEL prognosis is poor compared with that of HIV-associated diffuse large B-cell lymphoma (HIV-DLBCL) or Burkitt lymphoma.13,14 In the era of effective combination ART, published median overall survival remains <1 year.15 PEL has no established therapy but is sometimes treated similarly to other aggressive non-Hodgkin lymphomas with anthracycline-based regimens along with ART in HIV-infected patients. Durable complete remissions were noted in 43% of patients in the largest series treated with ART and CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone).
Factors associated with the clinical presentation and overall survival in PEL remain poorly characterized. The goals of the present study were to establish virologic, immunologic, and clinical features of PEL through comparison with 2 better-studied HIV-associated lymphoproliferative disorders, KSHV-MCD and HIV-DLBCL. We hypothesized that the inflammatory profile of patients with PEL is unique to KSHV-associated diseases and therefore distinct from that of patients with HIV-DLBCL, the most common lymphoma affecting people living with HIV. We further hypothesized that immune and/or viral biomarkers would have prognostic value in HIV-infected patients with PEL treated with ART and curative-intent modified EPOCH (infusional etoposide, vincristine, and doxorubicin with cyclophosphamide and prednisone).
Methods
Study design and patient selection
To evaluate baseline immunologic and clinical features in HIV-associated PEL, we performed a cross-sectional analysis comparing untreated patients with PEL vs HIV-infected patients with symptomatic KSHV-MCD or untreated HIV-DLBCL. We identified patients with these diseases participating in clinical studies (#NCT00092222, #NCT01419561, and #NCT00006518) in the HIV and AIDS Malignancy Branch and the Lymphoid Malignancies Branch clinics at the Center for Cancer Research, National Cancer Institute. All cases were pathologically confirmed in the Laboratory of Pathology, National Cancer Institute, and evaluated at the time of active disease. Diagnoses were based on cytopathology and/or hematoxylin and eosin staining of tissue supported by immunohistochemistry. KSHV tumor status was confirmed by staining for latency-associated nuclear antigen (anti-ORF73 rat monoclonal antibody; Advanced Biotechnologies, Eldersburg, MD). EBV tumor status in PEL was evaluated by in situ hybridization against EBV-encoded small RNA (EBER). Stored baseline serum specimens for cytokine analyses and peripheral blood mononuclear cells (PBMCs) for KSHV and EBV viral load were analyzed. Clinical records were reviewed for baseline clinical laboratory parameters before initiation of therapy. All lymphomas were staged according to the Lugano classification for non-Hodgkin lymphoma.16
To evaluate prognostic factors in PEL, retrospective survival analyses were performed in patients with PEL treated with ART and EPOCH modified by one or more therapies: rituximab, bevacizumab, and/or high-dose methotrexate. Patient records were evaluated for a history of other KSHV-associated diseases and clinical and laboratory abnormalities included in our working criteria for KICS. All patients signed informed consent on an institutional review board–approved protocol for correlative studies and biospecimen storage.
KSHV and EBV quantitative real-time polymerase chain reaction and KSHV serology
PBMC-associated KSHV and EBV viral DNA were quantified by using primers for KSHV K6 and EBV pol gene regions with cellular equivalents quantified by using human endogenous retrovirus 3 primers, as previously described,11 and reported as viral DNA copies per million PBMCs. KSHV serostatus of patients with HIV-DLBCL was determined by reactivity against K8.1 and/or latency-associated nuclear antigen using a bead-based multiplex Luminex assay.
Immunologic assays
Serum IL-6, IL-10, IL-1β, IL-8, IL-12 p70, interferon γ (IFN-γ), and tumor necrosis factor α (TNF-α) were evaluated by using a commercial multiplex assay (Meso Scale Discovery, Gaithersburg, MD). The Meso Scale IL-6 assay did not detect vIL-6 at concentrations up to 20 000 pg/mL. CD4+ T-cell counts were assessed by fluorescent-activated cell sorting. Plasma HIV-1 mRNA was measured by quantitative polymerase chain reaction using Amplicor HIV-1 Monitoring Kits (Roche Diagnostic Systems, Branchburg, NJ). HIV viral load was not measured in patients with HIV-DLBCL treated before the advent of combined ART in 1994. These patients were assumed to have HIV viral loads >100 copies/mL.
Statistical analysis
Clinical laboratory features and hypothesized inflammatory mediators of disease were compared between patients with PEL, KSHV-MCD, and HIV-DLBCL. Continuous parameters were compared between groups by using the exact 2-tailed Wilcoxon rank sum tests.
For survival analyses, cancer-specific mortality was evaluated from initiation of therapy to death with evidence of lymphoma or censoring at last follow-up in May 2016. Baseline factors were evaluated for prognostic value. These factors included history of KSHV-MCD and/or Kaposi sarcoma, tumor EBV status, and select clinical laboratory parameters: sodium, hemoglobin, platelets, albumin, C-reactive protein (CRP), lactate dehydrogenase, ferritin, CD4+ T-cell count, and HIV viral load. We also evaluated treatment-related factors, including protease inhibitor–based ART regimen, history of liposomal doxorubicin use, and inclusion of rituximab, bevacizumab, and/or high-dose methotrexate with EPOCH. The included immunologic and virologic biomarkers were those previously described as elevated in cases of KSHV-MCD or KICS: IFN-γ, TNF-α, IL-6, IL-8, IL-10, immunoglobulin E (IgE), ferritin, serum κ and λ free light chains, and KSHV and EBV viral load.17 Prognostic factors with continuous parameters were dichotomized near the median. Survival was evaluated by using the Kaplan-Meier method, and 2-sided log-rank tests were used to determine the statistical significance of differences between curves. Multivariable analyses used Cox regression with backward and stepwise selection, initially including variables with a univariate P value <.10. In view of the number of evaluations performed, a P value <.005 was considered statistically significant and between .005 and .05 was considered a strong trend.
Correlations between significant prognostic markers and EBV viral load were evaluated by using Spearman’s rank correlation. Correlation coefficients >0.7 were considered strong, between 0.5 and 0.7 moderately strong, between 0.3 and 0.5 weak to moderately strong, and <0.3 weak.
Results
Patient characteristics
We identified 20 HIV-positive patients with PEL diagnosed between 2000 and 2013: 19 men and 1 woman. Median age at diagnosis was 44 years. Eight patients had confirmed extracavitary disease in addition to cavitary disease, and 1 had extracavitary disease only. All patients had stage IV disease. Fifteen had concurrent Kaposi sarcoma, and 6 had concurrent KSHV-MCD. Nine patients had an HIV viral load <100 copies/mL, and median CD4+ count was 127 T cells/μL. Nineteen were treated with modified EPOCH and were included in the survival analyses. One patient included in the baseline cytokine analysis died before receiving treatment and was not included in this analysis. Tumor EBER status (EBV coinfection) was determined in these 19 patients; 14 were EBER positive and 5 were negative. Median potential follow-up time for PEL survival analyses was 5.9 years (range, 3.4-16.8 years). One patient was censored at the time of death from drug overdose with no evidence of PEL on autopsy 26 months after completing therapy. All other deaths were attributed to complications of PEL.
One comparison group of subjects comprised 20 patients with HIV-DLBCL diagnosed between 1983 and 2011. All patients with DLBCL had stage III or IV disease. Eighteen were men, and 2 were women with a median age of 39 years at diagnosis (range, 32-54 years). Median CD4+ count was 52 T cells/μL. The second comparison group comprised 19 HIV-infected patients with symptomatic KSHV-MCD diagnosed between 2001 and 2009: 17 men and 2 women with a median age of 43 years (range, 29-54 years) and median CD4+ count of 266 T cells/μL at diagnosis. No KSHV-MCD control patients developed PEL during follow-up. Tables 1 and 2 present complete patient characteristics.
Inflammatory milieu of PEL compared with KSHV-MCD and HIV-DLBCL
Nineteen patients with PEL had available baseline cytokine data and were included in the inflammatory milieu analysis. Eleven of the 14 patients with no history of KSHV-MCD also had complete CRP and KSHV data available for analysis, and all met criteria for KICS. Patients with PEL and patients with symptomatic KSHV-MCD without PEL had similar laboratory and inflammatory cytokine profiles, with no significant differences or strong trends toward differences in measured parameters. Sensitivity analyses, which excluded the 5 patients with PEL and concurrent KSHV-MCD, confirmed there were no differences between PEL patients without KSHV-MCD and the comparison patients with KSHV-MCD alone. By contrast, several clinical laboratory parameters and cytokines were found to be significantly different or with a strong trend toward difference between patients with PEL and comparison patients with HIV-DLBCL (Table 2). Despite a trend toward higher CD4+ counts (P = .021), patients with PEL had markedly elevated IL-10, hypoalbuminemia, and thrombocytopenia (P < .0001, .0027, and .0045, respectively). There were also strong trends toward differences between patients with PEL and HIV-DLBCL with respect to IL-1β, IL-6, and IL-12 (P = .019, .047, and .0093) (Figure 2). Sensitivity analysis restricted to patients with PEL and HIV-DLBCL with uncontrolled HIV viremia confirmed significant differences in IL-10 between the groups (P = .0027) and a strong trend toward clinically meaningful differences in hypoalbuminemia (P = .01) and thrombocytopenia (P = .05). PBMC-KSHV viral load was markedly elevated in PEL at levels comparable to that seen in KSHV-MCD. Three (15%) patients with HIV-DLBCL were KSHV seropositive, and only 1 patient had a detectable KSHV viral load. EBV viral load in PEL was comparable to that in KSHV-MCD and HIV-DLBCL, with no significant difference between groups.
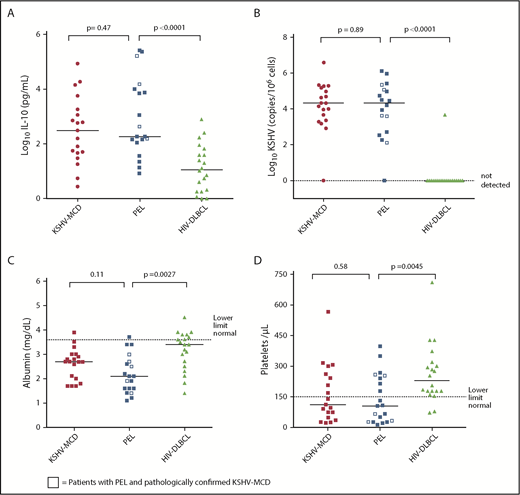
Clinical laboratory parameters and inflammatory cytokines in symptomatic PEL compared with KSHV-MCD and HIV-DLBCL. Between-group comparison of (A) log serum human IL-10 levels, (B) log KSHV viral loads, (C) albumin levels, and (D) platelet levels.
Clinical laboratory parameters and inflammatory cytokines in symptomatic PEL compared with KSHV-MCD and HIV-DLBCL. Between-group comparison of (A) log serum human IL-10 levels, (B) log KSHV viral loads, (C) albumin levels, and (D) platelet levels.
Curative-intent treatment in patients with PEL
Nineteen patients with PEL received modified EPOCH; the median number of cycles was 6. In addition to receiving EPOCH, 15 patients (80%) received rituximab, 9 (47%) received bevacizumab, and 3 (16%) received methotrexate. Six patients (32%) had previously received liposomal doxorubicin. All received ART, and in 8 patients (42%), this therapy included a protease inhibitor. Ten patients (53%) had a complete response to treatment.
Overall survival and factors associated with death
Median overall survival was 22 months, and 3-year cancer-specific survival was 47% (Figure 3). Baseline factors associated with inferior survival in univariate analysis were elevated serum IL-6 (P = .013), IL-10 (P = .041), and IgE (P = .019) levels (Table 3). Elevated ferritin (P = .054) and thrombocytopenia (P = .10) exhibited a tendency toward association with cancer-specific survival. In addition, median overall survival for EBER-positive and EBER-negative PEL cases was significantly different (P = .029). Median overall survival for EBER-positive cases was not reached, with a 3-year cancer-specific survival of 64%, whereas median overall survival in EBER-negative PEL was 10.4 months, with no patients alive at 3 years (Figure 3).

Cancer-specific survival in primary effusion lymphoma. (A) Cancer-specific survival in entire cohort, (B) according to tumor EBER status, (C) according to serum IL-6 above or below the median, and (D) according to serum IL-10 above or below the median. P values from 2-sided log-rank test. med, median.
Cancer-specific survival in primary effusion lymphoma. (A) Cancer-specific survival in entire cohort, (B) according to tumor EBER status, (C) according to serum IL-6 above or below the median, and (D) according to serum IL-10 above or below the median. P values from 2-sided log-rank test. med, median.
There were strong correlations among a number of inflammatory biomarkers in the patients with PEL (Table 4). IL-10 was moderately or strongly correlated with IL-6 and IFN-γ (r = 0.60, 0.76; P = .008 and .0002, respectively), and ferritin and platelets had a strong negative correlation (r = –0.82; P = .0002). Moderately strong correlations between IL-6 and ferritin (r = 0.59; P = .02), IL-10 and IgE (r = 0.54; P = .04), and KSHV and EBV viral load (r = 0.58; P = .009) were observed.
In Cox analyses with backward elimination starting with the significant factors observed in univariate analysis, IgE level was the strongest predictor of survival. Because of the uncertain significance of IgE in disease pathogenesis, 2 additional Cox models were created, both excluding IgE. In one, using backward elimination, EBER positivity was associated with significantly decreased risk of death (hazard ratio, 0.27; 95% CI, 0.076-0.93; P = .038), and in the other, using stepwise elimination, elevated IL-6 levels were associated with worse prognosis (hazard ratio, 6.12; 95% CI, 1.27-29.5; P = .024). Common clinical parameters such as hemoglobin, albumin, CRP, serum free light chains, lactate dehydrogenase, CD4+ count, HIV viremia, and treatment-related factors, as well as IL-8, TNF-α, IFN-γ, and KSHV and EBV viral loads, were not associated with overall survival (all P > .15). There was no significant difference in overall survival between patients with PEL and with and without KSHV-MCD (P = .39).
Discussion
Several important novel observations were noted in this study of 59 patients with HIV-associated lymphoproliferative disorders. First, there was significant clinical overlap between PEL, KSHV-MCD, and KICS. All patients with PEL with complete data had either concurrent KSHV-MCD or met working criteria for KICS. There were no significant differences in laboratory parameters or inflammatory cytokines between patients with PEL and symptomatic KSHV-MCD patients without PEL. Furthermore, patients with PEL with higher levels of inflammatory factors had a worse prognosis. Unexplained hypoalbuminemia and thrombocytopenia in HIV-infected patients with suspected lymphoma should raise the index of suspicion for PEL. We also noted that PEL in HIV occurs at a broader range of CD4+ T-cell counts than previously appreciated and can occur in the setting of effective HIV suppression. However, despite having higher CD4+ T-cell counts than the patients with HIV-DLBCL, patients with PEL had markedly elevated serum IL-10 levels and KSHV viral loads. By contrast, an elevated KSHV viral load was rarely detected in patients with HIV-DLBCL, even in KSHV-seropositive patients. KSHV viral load, therefore, may be helpful in the diagnostic evaluation of HIV-infected patients with suspected lymphoma. Overall, our findings show that cytokine dysregulation characterizes the unique pathogenesis, symptom profile, and natural history of PEL that distinguishes it from HIV-DLBCL. Our findings also identify for the first time that biomarkers of immune dysregulation are prognostic in patients with PEL.
KSHV-encoded oncogenes such as vIL-6 or viral homolog of the Fas-associated death domain-like IL-1-β-converting enzyme inhibitory protein (vFLIP) from PEL cells or other KSHV cellular reservoirs may contribute to pathogenesis.12 Previous research has shown that KSHV vIL-6 contributes to the pathogenesis of PEL, KSHV-MCD, and KICS and may be detected in effusions or serum in patients with these diseases.11,18,19 Transgenic mice with inducible vFLIP expression develop elevated IL-6, IL-10, and INF-γ levels, as observed in PEL, KSHV-MCD, and KICS.20 Lastly, PEL cells themselves may be a major source of IL-10, which is an autocrine growth factor in cell culture.19 Preclinical data suggest that KSHV may induce IL-10 expression through binding of the promoter region of the human IL-10 gene via KSHV replication and transcription factor in cooperation with host cellular transcription factors, and KSHV-encoded microRNA may induce secretion of IL-6 and IL-10 by macrophages and monocytes.21,22 Interestingly, EBV also encodes a viral homolog of IL-10 whose role in PEL is unknown.23
Hyperferritinemia, elevated IgE, and human cytokine profiles seen here in patients with PEL are associated with dysregulated cytokine activation (IL-10, IL-6, and IgE), KSHV replication, and macrophage activation syndromes (hyperferritinemia, thrombocytopenia, IFN-γ, IL-6, and IL-10). These findings suggest that abnormalities in both innate and acquired immunity are common in PEL and contribute to pathogenesis.15,24 Targeting cytokine dysregulation may have a role in future treatment approaches in PEL. In the treatment of KSHV-MCD, rituximab leads to rapid decreases in IL-6 and IL-10 levels, and this outcome was the rationale for its use in the treatment of patients with PEL in this series, despite PEL being a CD20– lymphoma. In PEL, rituximab was hypothesized to deplete the KSHV-infected B-cell reservoir and treat concurrent KSHV-MCD, which was present in 30% of the patients with PEL in this series. We are currently studying its use in PEL prospectively.
Another important and unexpected finding is that EBV status of the tumor is prognostic in PEL, with EBV positivity associated with improved survival. This finding is in contrast to HIV-DLBCL in which EBV positivity is associated with worse survival, suggesting that EBV plays different roles in the pathogenesis of PEL and HIV-DLBCL.25 Gene expression profiling in PEL effusions has shown that expression of cell cycle and signal transduction regulators differ between EBV-positive and EBV-negative tumors, suggesting they have distinct biologic features.26
To our knowledge, this study is the largest single-center cohort of patients with PEL treated with modified EPOCH backbone therapy in the combined ART era in the United States. A potential limitation was referral bias, as some of the cases had delayed diagnosis or treatment, which may have affected the patients with PEL, KSHV-MCD, and HIV-DLBCL differently. However, all PEL and HIV-DLBCL cases were stage III or IV, and significant differences in cytokine profile were still noted between the 2 groups. In addition, a greater proportion of HIV-DLBCL cases had uncontrolled HIV, and conclusions regarding relative CD4+ T-cell counts are therefore not generalizable in the ART era. Previous studies have shown that HIV viremia and CD4+ lymphocytopenia tend to lead to increased inflammatory cytokines and worse cytopenias than when HIV viremia is controlled. These significant differences in inflammatory cytokines and clinical features, despite potential confounding of HIV and CD4+ T-cell counts, suggest that the distinct clinical presentation of PEL is largely a consequence of KSHV-associated pathogenesis rather than a sequela of untreated HIV.12 These findings are further supported by the lack of observed differences between PEL and KSHV-MCD in these same clinical, immunologic, and viral parameters.27,28 Lastly, our sample size was able to identify PEL-associated prognostic factors with strong effect sizes. Larger studies are needed to identify additional factors and for comparisons with other rarer HIV-associated lymphoma subtypes.
Importantly, the present study provides further evidence that PEL should be approached with curative intent. In our series, modified EPOCH resulted in 3-year cancer-specific survival of 47%. No patient who survived 2 years after the diagnosis of PEL has subsequently died of PEL. Despite improvement over previously reported outcomes, the addition of agents targeting KSHV-driven oncogenesis would likely further improve survival.29 Lenalidomide, a drug with multiple immune modulatory targets, including KSHV-driven IFN response factor 4 upregulation, has been shown to be cytotoxic in PEL tumor lines and reverse KSHV-induced downregulation of major histocompatibility complex-1, B7-2 and ICAM-1.20,30-32 A clinical study of lenalidomide combined with EPOCH and rituximab in patients with PEL is currently evaluating this approach (#NCT02911142).
Presented in part in abstract form at the 54th annual meeting of the American Society of Hematology, Atlanta, GA, 9 December 2012; the 56th annual meeting of the American Society of Hematology, San Francisco, CA, 7 December 2014; the 16th International Conference on Malignancies in HIV/AIDS, Bethesda, MD, 23-24 October 2017; and the International Conference on EBV & KSHV, Madison, WI, 28 July-4 August 2018.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Kirsta Waldon for patient and clinical care coordination, Adam Rupert for assistance with cytokine analyses, and Giovanna Tosato for discussions regarding the pathogenesis of primary effusion lymphoma.
This research was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health, by US federal funds from the National Cancer Institute, National Institutes of Health (HHSN261200800001E), and ZIA BC011700 (T.S.U.).
Authorship
Contribution: T.S.U., K.L., and R.Y. designed the study. K.L., T.S.U., M.N.P., K.A., M.B., R.R., K.M.W., P.H.G., R.F.L., W.W., and R.Y. cared for patients. A.C.F., S.P., and E.S.J. provided pathologic confirmation of case subjects and control subjects. D.W. designed and V.A.M. and W.M. conducted the KSHV and EBV assays. K.L., T.S.U., and M.B. collected data. K.L., T.S.U., M.B., S.M.S., and R.Y. analyzed data. All authors contributed to writing and approving the manuscript.
Conflict-of-interest disclosure: T.S.U., M.N.P., and R.Y. are coinventors on a patent related to the treatment of KSHV-associated diseases with immune modulatory compounds; R.Y. is also coinventor on other patents not related to the current study, and his spouse is a coinventor on a number of patents, including one on the measurement of KSHV vIL-6. These inventions were all made as part of their duties as employees of the US Government, and the patents are or will be assigned to US Department of Health and Human Services. The government may convey a portion of the royalties it receives from licensure of its patents to its employee inventors. R.Y. and T.S.U. have a Cooperative Research and Development Agreement with Celgene Corp. and recently conducted clinical research using drugs supplied to the National Cancer Institute by Merck & Co., Hoffmann-La Roche, and Bayer Healthcare. R.Y. has a material Cooperative Research and Development Agreement with CTI BioPharma Corp. for the use of their drug in the laboratory. The remaining authors declare no competing financial interests.
Correspondence: Thomas S. Uldrick, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, Mail Stop M1-B140, Seattle, WA 98109; e-mail: tuldrick@fredhutch.org.


