

Ibrutinib works, but does using it set patients up for potential relapse by causing increased genomic instability? This question is addressed in this issue of Blood by , who provide convincing evidence that resolves a key safety issue surrounding the use of this drug, giving new insight into its mechanism of action.1
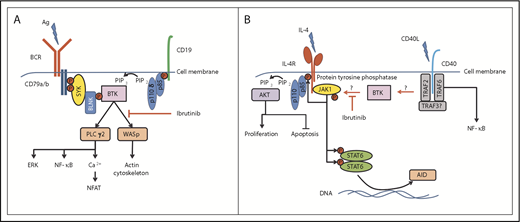
Central role of BTK in the regulation of BCR- and CD40L/IL-4–induced signaling. (A) BTK is centrally involved in the BCR-signaling pathway, sitting distal to Syk and PI3Kδ, to phosphorylate and activate PLCγ2 and WASp, leading to further signal pathway activation and actin cytoskeleton rearrangements, respectively. (B) The mechanism of how BTK is activated by CD40 ligation is unknown, but could involve PI3K isoforms other than PI3Kδ (involved in BCR signaling). Active BTK then potentially interacts with a protein tyrosine phosphatase, which subsequently dephosphorylates JAK1 and STAT6 to deactivate their function in promoting cell proliferation (through the PI3K pathway) and induction of AID expression. Ag, antigen; BLNK, B-cell linker protein; ERK, extracellular signal-regulated kinase; IL-4R, IL-4 receptor; NFAT, nuclear factor of activated T cells; P, phosphorylated tyrosine motif; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; TRAF, tumor necrosis factor receptor–associated factor.
Central role of BTK in the regulation of BCR- and CD40L/IL-4–induced signaling. (A) BTK is centrally involved in the BCR-signaling pathway, sitting distal to Syk and PI3Kδ, to phosphorylate and activate PLCγ2 and WASp, leading to further signal pathway activation and actin cytoskeleton rearrangements, respectively. (B) The mechanism of how BTK is activated by CD40 ligation is unknown, but could involve PI3K isoforms other than PI3Kδ (involved in BCR signaling). Active BTK then potentially interacts with a protein tyrosine phosphatase, which subsequently dephosphorylates JAK1 and STAT6 to deactivate their function in promoting cell proliferation (through the PI3K pathway) and induction of AID expression. Ag, antigen; BLNK, B-cell linker protein; ERK, extracellular signal-regulated kinase; IL-4R, IL-4 receptor; NFAT, nuclear factor of activated T cells; P, phosphorylated tyrosine motif; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; TRAF, tumor necrosis factor receptor–associated factor.
Ibrutinib is increasingly becoming a standard of care in the treatment of chronic lymphocytic leukemia (CLL); recent studies of its long-term use have shown both sustained efficacy and acceptable tolerability.2,3 Importantly, approximately one-third of CLL patients relapse on ibrutinib therapy, and disease resistance in many of these cases is attributed to mutations in either Bruton tyrosine kinase (BTK), the target of ibrutinib, or phospholipase Cγ2 (PLCγ2), a principal target of BTK within B cells.4 Worryingly, a preclinical study by Compagno et al5 suggested that inhibition of BTK increased genomic instability in normal and neoplastic B cells via a mechanism involving enhanced expression of activation-induced cytidine deaminase (AID). The implication here is that ibrutinib treatment may predestine CLL patients to drug resistance by causing increased genomic instability and consequent mutation in BTK and PLCγ2. However, the findings of Compagno et al were derived using MEC-1 cells, a CLL cell line descended from the malignant cells of a patient in prolymphocytoid transformation.6 The same effect of ibrutinib in primary CLL cells may not be the case, and these are exactly the data Morande et al present. Using a combination of observations gained from primary CLL cells in in vivo and in vitro settings, these investigators find that ibrutinib treatment not only reduces levels of AID in cells from patients enrolled in a clinical trial, but also inhibits induction of AID expression in primary CLL cells that are being stimulated with CD40L and interleukin 4 (IL-4). This latter finding is important because CD40L/IL-4 is thought to be a key microenvironmental factor contributing to malignant clone survival and proliferation in CLL. When coupled to the former finding that ibrutinib limits AID expression in vivo, there is a strong suggestion that genomic instability caused by microenvironmental influence can be prevented by this drug. Thus, the worry that ibrutinib may sow the seeds of its demise seems to be unfounded.
The role of microenvironment in CLL cannot be understated. By playing a role in survival and proliferation of the malignant cells in this disease, microenvironmental influences contribute majorly to the expansion of clones that are resistant to therapy. Within this context, ibrutinib targets BTK to simultaneously cause redistribution lymphocytosis by impeding the ability of CLL cells to reside within proliferation centers, and blocking the ability of these cells to respond to chemokines that stimulate entry/reentry to this environment.7 This effect on decreasing tumor burden is augmented by the reduced proliferation and enhanced death of CLL cells that is observed in patients being treated with ibrutinib.8 Thus, our current understanding of ibrutinib’s therapeutic effect is that it is mediated by removing CLL cells from proliferation centers and reducing their overall survival. Morande et al add to this understanding by measuring AID expression in CLL cells that are Ki-67+, and showing that this population of cells is severely reduced after patients begin taking ibrutinib. This means that ibrutinib treatment blocks the means of creating mutation within CLL cells, as well as the means through which clones bearing mutation can be expanded. So why do patients on ibrutinib develop resistance, particularly that associated with mutation of BTK and PLCγ2? The answer to this is not clear but could be related to the amount of treatment received by a given patient prior to ibrutinib therapy. Such patients are particularly prone to developing ibrutinib resistance,4 likely from clones already containing mutant BTK and PLCγ2 that are reported present at low frequency9 rather than through de novo mutagenesis.
So how does ibrutinib affect CLL cells in patients? This compound has a high degree of specificity for BTK, and the main downstream targets of this tyrosine kinase in B cells are PLCγ2 and Wiskott-Aldrich syndrome protein (WASp), but this might not be all it does. To more fully understand how ibrutinib therapy impacts CLL cells, Morande et al isolated malignant cells bearing a phenotype associated with having a high proliferative index from CLL patients enrolled in a clinical trial before and during receipt of the drug. They then performed a phosphoprotein analysis on these cells and identified that ibrutinib treatment leads to dephosphorylation of JAK1 at Tyr1022, an event that deactivates the function of this kinase. Active JAK1 in cells stimulates the phosphatidylinositol 3-kinase (PI3K) pathway; consistent with its deactivation in ibrutinib-treated CLL cells, the authors observe changes in the phosphorylation of key proteins connected with this pathway that function in controlling proliferation and apoptosis. This could possibly account for the reduction of cells expressing Ki-67 and the increase in CLL cell death that is observed in these patients. The explanation for reduced AID expression is that JAK1 targets STAT6 for phosphorylation, which then translocates to the cell nucleus and is responsible for induction of AID expression. Indeed, Morande et al model this in vitro and show that ibrutinib treatment of CD40L/IL-4–stimulated CLL cells reduces STAT6 phosphorylation and AID expression. Thus, a potentially new role for BTK can be applied that is different to its typical role in the B-cell receptor (BCR)–signaling pathway (see figure panel A). In this new role, BTK is key to modulating JAK1 activation within CLL cells stationed within the microenvironment and exposed to CD40L/IL-4 (see figure panel B). It is known that BTK becomes activated in CD40-stimulated B cells,10 and, within this context, there may be new substrates, such as protein tyrosine phosphatases, that are able to dephosphorylate JAK1 and STAT6 and downregulate IL-4 signaling. Determining this will require further experiments comparing the effect of ibrutinib on CLL cells incubated with IL-4 alone and with CD40L/IL-4.
The article by Morande et al therefore sets the stage for further work into the mechanism of action of ibrutinib and similar compounds. These compounds can no longer be referred to as just BCR-signaling pathway inhibitors as they seemingly also affect a key pathway responsible for the development and expansion of resistant clones. Thus, ibrutinib is safer than we thought and may be most useful as a frontline therapy.
Conflict-of-interest disclosure: J.R.S. received funding from Verastem Inc., and has a collaborative relationship with the Netherlands Translational Research Center B.V.

