

In this issue of Blood, describe a novel CRISPR/Cas approach for correcting β-thalassemias that result from aberrant donor or acceptor splice sites.1 The authors designed guide RNA/Cas complexes that can be introduced efficiently into hematopoietic stem and progenitor cells (HSPCs) ex vivo without viral vectors and can create double-stranded DNA cuts at specific genomic sites. The cells rapidly repair these double-stranded breaks by nonhomologous end-joining, which typically results in small deletions around the cut site. If these deletions encompass the aberrant splice sites and avoid additional sequences necessary for efficient splicing (see figure), normal splicing can be restored.
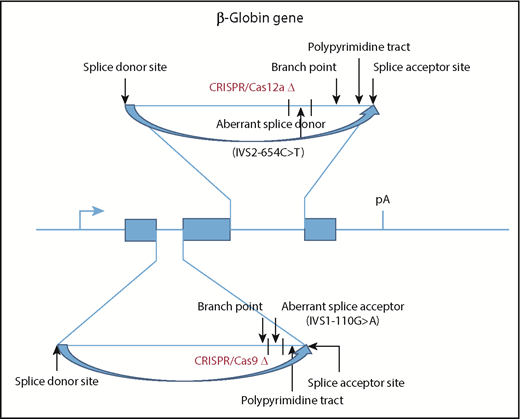
CRISPR/Cas deletion of aberrant splice sites provides a novel therapeutic approach for β-thalassemia. Two prevalent β-thalassemia mutations are illustrated in the human β-globin gene: IVS2-654C>T, which produces an aberrant splice donor site in IVS2, and IVS1-110G>A, which produces an aberrant splice acceptor site in IVS1. Additional sequences necessary for efficient splicing are also indicated. Xu et al demonstrate that electroporation of CD34+ HSPCs with ribonucleoproteins composed of Cas9 or Cas12a protein and guide RNAs complementary to the IVS1 or IVS2 mutations significantly increase the level of correctly spliced β-globin mRNA and, consequently, the amount of HbA produced in red blood cells after in vitro differentiation.
CRISPR/Cas deletion of aberrant splice sites provides a novel therapeutic approach for β-thalassemia. Two prevalent β-thalassemia mutations are illustrated in the human β-globin gene: IVS2-654C>T, which produces an aberrant splice donor site in IVS2, and IVS1-110G>A, which produces an aberrant splice acceptor site in IVS1. Additional sequences necessary for efficient splicing are also indicated. Xu et al demonstrate that electroporation of CD34+ HSPCs with ribonucleoproteins composed of Cas9 or Cas12a protein and guide RNAs complementary to the IVS1 or IVS2 mutations significantly increase the level of correctly spliced β-globin mRNA and, consequently, the amount of HbA produced in red blood cells after in vitro differentiation.
Hemoglobinopathies are the most common hereditary disorders worldwide, and new therapeutic approaches are urgently needed. Lentiviral-mediated gene addition therapies have improved tremendously in the past few years, and patients with sickle cell disease and β-thalassemia have recently been cured. This is great news for patients and their families. However, there are significant concerns about the cost of viral-mediated gene therapy, so additional approaches are being investigated. Gene editing methods using purified zinc finger proteins, transcription activator-like effector nucleases, and CRISPR/Cas complexes provide biochemical approaches that could substantially reduce the cost of therapy and may provide more genome site specificity.
Xu et al convincingly demonstrate that CRISPR/Cas-mediated deletion of aberrant splice sites in blood CD34+ HSPCs from β-thalassemic patients significantly increases the level of correctly spliced β-globin messenger RNA (mRNA) and, consequently, the amount of adult hemoglobin (HbA) in erythroid cells after in vitro differentiation. Perhaps the most impressive results are illustrated in Figure 2F from the accompanying article by Xu et al for β+/β0-thalassemic cells (partial/complete loss of β function) and β+/βE-thalassemic cells (partial/mild loss of β function) in which the level of HbA is increased from an average of 9.9% of total hemoglobin to 59.1%. The only caveat is that the total amount of hemoglobin (normally 29 ± 2 pg per cell) was not measured; therefore, it is difficult to know whether hemoglobin levels were normalized. This is important because the level of hemoglobin measured in grams per deciliter of blood can be as low as 2 g/dL in β+/β0-thalassemia patients, and 8 to 10 g/dL is required to render a patient transfusion independent. The good news is that the in vitro differentiation data for β+/β0-thalassemia patients #4 and #5 in the Xu et al article suggest that a sufficient level of total hemoglobin may have been obtained. If the corrected HSPCs were transplanted into patients, these individuals might be cured.
CRISPR/Cas deletion of aberrant splice sites has also recently been described for severe retinal dystrophy.2 The Xu et al article and the article by Maeder et al2 demonstrate that this novel strategy may be close to clinical application, and again, this is great news for patients and their families.
Conflict-of-interest disclosure: T.M.T. is a shareholder in HemEdits, LLC.

