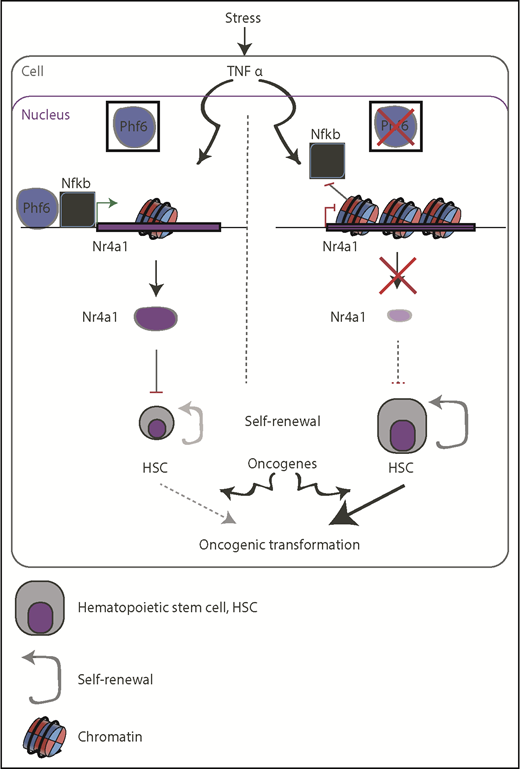
In the current issue of Blood, investigated the role of the plant homeodomain-like finger protein 6 (PHF6) in hematopoietic stem cell (HSC) self-renewal in an attempt to further clarify the previously suggested tumor suppressive role of this protein in human leukemia.1 Interestingly, Phf6 ablation rendered HSCs less responsive to inflammatory stress signals, but was able to increase HSCs proliferation rates, which might, at least in part, contribute to oncogenic transformation (see figure).
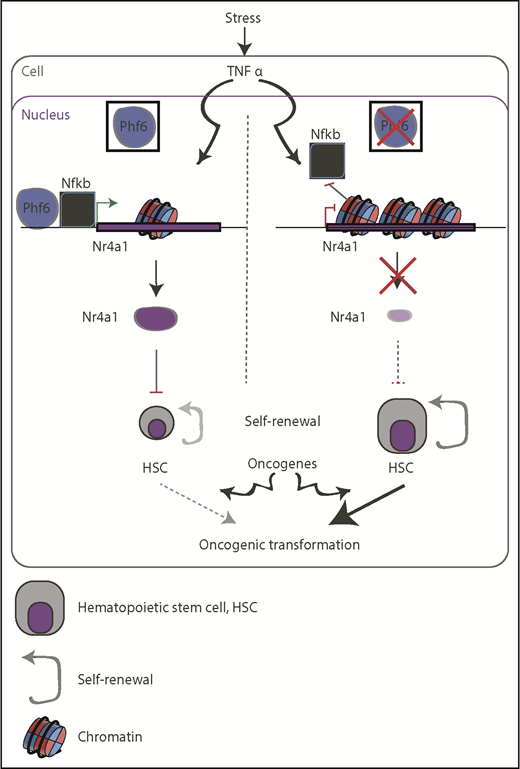
Schematic representation of the suggested function of PHF6 in restricting the self-renewal capacity of HSCs (left) and how it can go awry in PHF6-mutant backgrounds (right). Lack of PHF6 could be exploited by oncogenic transcription factors, such as NOTCH1 and TLX3, to promote oncogenesis (bottom).
Schematic representation of the suggested function of PHF6 in restricting the self-renewal capacity of HSCs (left) and how it can go awry in PHF6-mutant backgrounds (right). Lack of PHF6 could be exploited by oncogenic transcription factors, such as NOTCH1 and TLX3, to promote oncogenesis (bottom).
PHF6 is located on the X chromosome, and children with mutations in PHF6 develop a hereditary X-linked disorder presenting with mental retardation and physical deformities, called Borjeson-Forssman-Lehmann syndrome (BFLS).2 Loss-of-function mutations or deletions targeting PHF6 have been initially identified by the Ferrando group in ∼30% of T-cell acute lymphoblastic leukemia (T-ALL) cases.3 In line with this notion, 1 case report described the development of T-ALL in a child who suffered from BFLS. Nevertheless, the functional role of PHF6 as a tumor suppressor gene in human leukemia remains largely elusive.
PHF6 is chromatin-bound protein that recognizes histone methylation marks through its 2 PHD-like zinc finger domains, but it lacks enzymatic activity and it might play structural roles in different protein complexes. In line with this notion, it has recently been shown that PHF6 can bind the chromatin remodeling complex nucleosome remodeling and deacetylase (NURD), potentially controlling p21 gene and response to glucocorticoids in T-ALL.4 In addition, it was postulated that PHF6 also participates in PAF1-associated transcriptional elongation (see Todd et al, for a review of those functions5 ). PHF6 also binds the upstream binding factor 1 via the first PHD domain and suppresses ribosomal RNA transcription in the nucleolus to control cell cycle.5 Furthermore, recent studies showed that PHF6 binds to additional proteins, such as the BRWD2/PHIP protein, that controls gene expression and cross talk of histone acetylation and methylation6 on enhancers. Characterization of the exact role of PHF6 in those complexes is work in progress.
Using deletion of exons 4 and 5 of the gene, which encode the second plant homeodomain of Phf6, the authors showed that loss of functional Phf6 renders HSCs (the CD150+ CD34− lin− sca/c-kit+ population) less sensitive to inflammatory cytokines or other stresses. The authors also interrogated the roles of Phf6 in embryonic hematopoiesis using bone marrow material from knockout and wild-type animals in a series of mouse transplantation studies. This analysis revealed that Phf6 knockout cells are more capable of reconstituting the hematopoietic system than their wild-type counterparts. Although PHF6 mutations are quite prevalent in adult T-ALL, adult mouse hematopoietic reconstitution only slightly differed between the wild-type and mutant Phf6 backgrounds in the absence of stress, such as transplantation, chemotherapy, or inflammation.
Next, the authors sought out to dissect the mechanistic underpinnings of PHF6 function. They observed significant differences in chromatin accessibility between CD34+/CD150− HSCs from control and Phf6 mutant animals, in agreement with participation of PHF6 in the NURD complex. Moreover, the authors showed that tumor necrosis factor α (TNFα), transforming growth factor-β, and apoptosis pathways are downregulated in Phf6-mutant HSCs. At the same time, E2F pathway, a positive controller of cell cycle, is upregulated in Phf6-deleted HSCs, which might explain the proliferative phenotype of the Phf6 null HSCs.
As TNFα restricts the self-renewal ability of HSCs, Miyagi et al also interrogated the ability of TNFα to restrict HSC proliferation in the presence and absence of Phf6. Indeed, Phf6 mutant HSCs were largely insensitive to TNFα as compared with wild-type cells. Chromatin immunoprecipitation and accessibility studies showed that Phf6 binds and relaxes the chromatin in loci-targets of the TNFα pathway, potentially allowing binding of nuclear factor κB (Nfkb) protein and activation of Nr4a1 gene. This gene codes for a ligand-independent nuclear receptor that can activate downstream target genes, such as Junb, that restrict HSC proliferation.
Recent molecular studies from the Ferrando group used in vivo mouse studies and HSCs from a different Cre recombinase deleter strain to also show regulation of HSC self-renewal via Phf6, albeit with different proposed mechanistic underpinnings.7 In this case, the authors demonstrate a potentially critical upregulation of the JAK/STAT pathway and MYC targets, which could be exploited as a therapeutic vulnerability in PHF6 mutant leukemia. They also showed that Phf6 deletion lowers the threshold for oncogenic transformation in a mouse model of NOTCH1-induced T-ALL. Intriguingly, PHF6 and JAK1 mutations coexist in T-ALL. Similarly, McRae et al showed that Phf6 loss potentiates the proliferative phenotype of stem cells and the oncogenic ability of T-cell leukemia homeobox 3 (TLX3, a T-ALL oncogene) expression in mice.8
Cancer stem cells are considered the cornerstone of therapy resistance, and these new findings associate PHF6 function with stem cell homeostasis, which might pave the pathway for further studies using leukemia stem cells. Further studies are warranted to (a) address the role of PHF6 in adult compared with pediatric disease, and in different types of cancer as well as stresses such as inflammation or transplantation; and (b) identify therapies for PHF6-mutant cancers. Regarding (a), it is known that PHF6 is a tumor suppressor in acute myeloid leukemia (AML; ∼3% of AML cases have PHF6 mutations5,9 ), whereas it presents with no mutations in B-cell acute lymphoblastic leukemia (B-ALL) and acts as an oncogene in in vivo contexts of B-ALL.10 As Nr4a proteins control cell cycle in AML contexts, one would expect that similar mechanisms could be shared between leukemias with PHF6 mutations. Regarding (b), understanding PHF6 co-recruitment to chromatin with different transcription factors and participation in different transcriptional complexes in different contexts is critical toward the development of therapeutic interventions. For instance, the role of PHF6 in the NF-κB transcriptional program should be further interrogated in normal and leukemia systems especially as the NF-κB pathway is activated by NOTCH1 in T-ALL contexts, which might complicate the role of PHF6 as an NF-κB partner. These recent findings pave the way for the use of JAK/STAT inhibitors or epigenetic inhibitors, such as the ones against bromodomain-containing proteins (found to inhibit the activity of the MYC pathway) in leukemia with PHF6 mutations.
Conflict-of-interest disclosure: The author declares no competing financial interests.