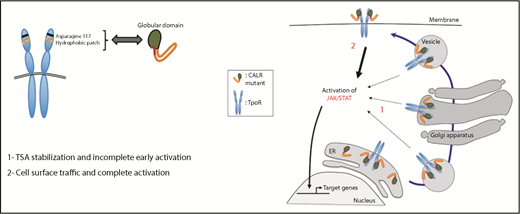
The discovery by exome sequencing that calreticulin (CALR) gene mutations are important gain-of-function myeloproliferative neoplasm (MPN) driver mutations1,2 was as inexplicable as it was unexpected. In this issue of Blood, 3 report that mutated CALR behaves like a rogue chaperone, usurping the role of JAK2 and promiscuously transporting immature or traffic-defective thrombopoietin receptors (TpoRs) to the cell surface from the endoplasmic reticulum (ER) (see figure).
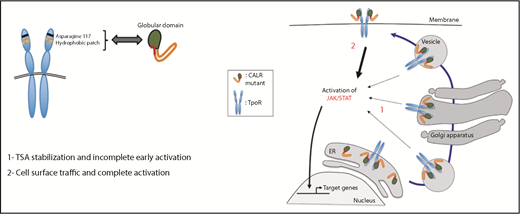
Diagram of the principle features of the mutated CALR–TpoR interaction leading to cell surface transport from the ER, TpoR activation, and signal transduction. See Figure 6E in the article by Pecquet et al that begins on page 2669.
Diagram of the principle features of the mutated CALR–TpoR interaction leading to cell surface transport from the ER, TpoR activation, and signal transduction. See Figure 6E in the article by Pecquet et al that begins on page 2669.
The MPNs, polycythemia vera, essential thrombocytosis, and primary myelofibrosis, are clonal hematopoietic stem cell (HSC) disorders because all MPN driver mutations are expressed in HSCs. MPN driver mutations directly or indirectly activate JAK2, the cognate tyrosine kinase of the erythropoietin receptor (EpoR) and the TpoR (also referred to as MPL). JAK2 can also be used by the granulocyte colony-stimulating factor (G-CSF) receptor, which accounts for the shared clinical features of the MPNs.
JAK2, however, is not only a tyrosine kinase, it is a chaperone for the EpoR and the TpoR. Specifically, with the TpoR, JAK2 enhances its stability, its cell surface presence, and its total cellular concentration.4 The EpoR and TpoR also differ in other important ways. EpoRs are transported to the cell surface fully mature; TpoRs are transported to the cell surface in immature and mature conformations, both of which engage in signal transduction.
EpoRs are downregulated from the cell surface and degraded after signal transduction by proteasomal and lysosomal pathways. TpoRs recycle to the cell surface after signal transduction and downregulation, presumably because they are essential for HSC maintenance, as well as for hematopoietic progenitor cell proliferation. TpoRs also have a reduplicated cytokine receptor homology domain (CRHD), where thrombopoietin (Tpo) and mutated CALR bind, and which is a hot spot for inherited mutations causing thrombocytosis or thrombocytopenia. Because TpoR is the only hematopoietic growth factor receptor in HSCs, MPNs are functionally thrombopoietin receptor disorders.
Calreticulin is a multifunctional soluble ER-resident protein that is responsible for folding and glycosylation of nascent glycoproteins and ER calcium homeostasis; although wild-type CALR has functions outside the ER,5 it is not a protein chaperone and does not bind wild-type TpoR or TpoR mutants. Structurally, CALR has an N-terminal ER-localizing signal sequence, an N-terminal lectin-(glycan) binding domain, a P domain with high-affinity Ca2+ binding sites, and a negatively charged C-terminal tail with low-affinity Ca2+ binding sites and a KDEL ER-retention sequence.
CALR driver mutations, all in exon 9, include a 52-base deletion (designated type 1) and a 5-base insertion (designated type 2), both of which cause a +1-base frameshift, resulting in a positively charged C-terminal tail (shorter in the type 1 mutation) lacking the KDEL ER-retention sequence.1,2
Previous studies demonstrated that the lectin-binding domain and the neomorphic positively charged C-terminal tail of mutated CALR are essential for TpoR binding and activation6,7 ; that mutated CALR must bind to the TpoR distal extracellular CRHD as a homomultimer for JAK2 activation8 ; that mutated CALR binds strongly to the TpoR, weakly to the G-CSF receptor, and not at all to the EpoR9 ; and, finally, that cell surface expression of the mutated CALR–TpoR complex is mandatory for TpoR activation and JAK2 signaling6 (see figure). By itself, mutated CALR has no oncogenic or paracrine activity.
Pecquet et al now demonstrate that the TpoR contains a hydrophobic patch in its distal CRHD, which is essential for mutated CALR binding and is absent in the EpoR, explaining the lack of EpoR activation by the mutant protein. They also demonstrate that mutant CALR can transport incompletely glycosylated wild-type TpoR from the ER through the Golgi apparatus to the cell surface, as well as rescue from ER retention the TpoR MPLR102P, which causes congenital amegakaryocytic thrombocytopenia (CAMT), as well as other ER traffic-defective TpoRs.
Importantly, wild-type TpoR bound to mutated CALR could still respond to Tpo, the plasma level of which is elevated in the MPN, whereas the TpoR MPLR102P, once rescued, could respond to a Tpo mimetic.
The report by Pecquet and colleagues suggests, first, that attention currently focused on inhibiting the ER chaperone function of JAK2V617F might be profitably engaged on inhibiting the chaperone function of mutated CALR. Second, the observation that the TpoR MPLR102P, when expressed at the cell surface, can be stimulated by a Tpo mimetic implies that, in the future, CALR gene editing might be a useful strategy for correcting CAMT. Finally, the expression of a neoantigen, mutated CALR, on the surface of MPN HSCs provides a rationale for immunotherapy in CALR mutation–positive MPN patients.1
Unsurprisingly, interesting experimental observations lead to new inquiries. In this instance, such inquiries would include the mechanism for wild-type JAK2 activation by mutated CALR, how wild-type JAK2 activated by mutated CALR avoids physiologic inhibition, and the length of time that mutated CALR–TpoR complexes reside on the cell surface. Normally, Tpo-induced JAK2 activation is accompanied by upregulation of SOCS3 and LNK, which attenuate JAK2 activity but not TpoR recycling. JAK2V617F paradoxically abbreviates the cell surface residence of mature TpoR by ubiquitination and blocks TpoR recycling,10 but it remains to be determined whether mutated CALR mimics JAK2V617F in this regard.
Conflict-of-interest disclosure: The author declares no competing financial interests.