Key Points
Mass spectrometry–based proteomics is a powerful technique that can identify IGVL gene products from tissue specimens.
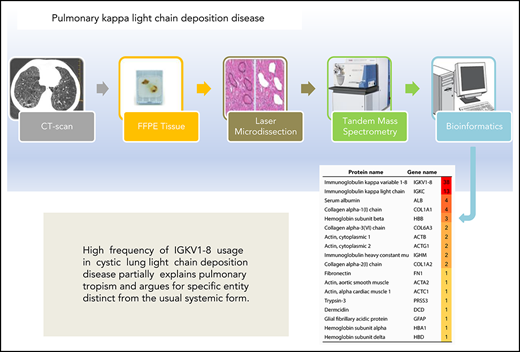
High frequency of IGKV1-8 usage in cystic lung LCDD partially explains pulmonary tropism and argues for a distinct entity.

Abstract
We previously reported a new form of light chain deposition disease (LCDD) presenting as diffuse cystic lung disorder that differs from the usual systemic form with respect to patient age, the male/female ratio, the involved organs, and the hematologic characteristics. We also demonstrated that the light chains were produced by an intrapulmonary B-cell clone and that this clone shared a stereotyped antigen receptor IGHV4-34/IGKV1. However, we only analyzed 3 patients. We conducted a retrospective study including lung tissue samples from 24 patients with pulmonary LCDD (pLCDD) matched with samples from 13 patients with pulmonary κ light chain amyloidosis (pAL amyloidosis) used as controls. Mass spectrometry–based proteomics identified immunoglobulin κ peptides as the main protein component of the tissue deposits in all patients. Interestingly, in pLCDD, IGKV1 was the most common κ family detected (86.4%), and IGKV1-8 was overrepresented compared with pAL amyloidosis (75% vs 11.1%, P = .0033). Furthermore, IGKV1-8 was predominantly associated with a diffuse cystic pattern (94%) in pLCDD. In conclusion, the high frequency of IGKV1-8 usage in cystic pLCDD constitutes an additional feature arguing for a specific entity distinct from the systemic form that preferentially uses IGKV4-1.
Introduction
Light chain deposition disease (LCDD) is a rare disorder related to the accumulation of monoclonal light chains in basement membranes.1 In contrast to amyloidosis, the deposits are Congo red negative and nonfibrillar ultrastructurally.1 In 2006, 30 years after the initial description of the systemic form by Randall, we reported a new form of LCDD presenting as diffuse cystic lung disorder that, in the most severe cases, may require lung transplantation.2 It differs from the systemic form with respect to the age at diagnosis, the male/female ratio, the involved organs, and the hematologic characteristics.2,3 Indeed, it occurs predominantly in younger patients, primarily women, does not affect the kidney, the liver, or the heart, and is frequently, but not always, associated with an abnormal serum-free light chain ratio.2-5 Despite the lack of morphological criteria for a pulmonary B-cell neoplasm, we demonstrated, using polymerase chain reaction, that the light chains were produced by an intrapulmonary B-cell clone.6 We also found that this clonal expansion shared an unmutated antigen receptor IGHV4-34/IGKV1 with heavy and light chain CDR3 sequences of >80% amino acid identity.6 However, we analyzed only 3 patients.6 Since this initial report, we have not identified evidence in subsequent patients against the pulmonary origin of κ light chain production: examination of the bone marrow biopsy/aspiration was always normal, there was no systemic involvement, an intrapulmonary B-cell clone was found, and the recurrence of the disease after lung transplantation was very rare, even without any complementary biotherapy/chemotherapy.
In the past few years, mass spectrometry (MS)-based proteomics has become the gold-standard technique for the classification of amyloidosis from tissue samples.7,8 We have established that this technique is also a reliable tool for the diagnosis of pulmonary LCDD (pLCDD).9 The use of this method has been expanded to the identification of the peptides derived from IGVL genes and gene family with 100% specificity.10,11 Therefore, we designed the present study to investigate IGVL gene usage in a cohort of pLCDD to provide clues about the molecular characteristics of this peculiar disorder.
Study design
Patient population and controls
We conducted a retrospective study including lung samples from 24 patients with localized pLCDD, identified from 2004 to 2018, retrieved from our collection. Among them, 10 have already been published.2,3,6,9 The samples from 13 patients with pulmonary κ light chain amyloidosis (pAL amyloidosis) identified during the same period served as controls (12 localized forms and 1 systemic form). The diagnosis of LCDD and AL amyloidosis was established based on biopsy specimens or explanted lungs using Congo red, immunofluorescence, electron microscopy, and/or MS-based proteomics. Based on computed tomography, the patients were categorized as having diffuse cystic disorder, solitary nodule, bronchial and bronchiolar involvement, or diffuse interstitial disease without cysts. Patient consent was obtained according to the Institutional Review Board of CHU de Toulouse.
MS-based proteomic analysis
Ten-micrometer-thick sections of formalin-fixed paraffin-embedded tissue were mounted on slides (Expression Pathology) and stained with hematoxylin and eosin. One hundred thousand square micrometers of deposits were selected by laser microdissection (Leica 6500; Leica Microsystems). Proteins were extracted from the collected material in ammonium bicarbonate buffer, reduced with dithiothreitol, and alkylated with iodoacetamide. Then, proteins were digested into peptides with trypsin (Sigma) and analyzed by nanoscale liquid chromatography (LC), coupled with tandem MS (MS/MS), using an UltiMate 3000 RSLCnano System (Dionex) coupled to an Linear Trap Quadripole Orbitrap Velos mass spectrometer (Thermo Fischer Scientific). Data were processed with Mascot (version 2.5.2) against human entries in the Swiss-Prot protein database augmented with known protein sequences from human IGVL genes obtained from the ImMunoGeneTics database. Validation of results was performed through a false-discovery rate set to 1% at protein and peptide-sequence match levels determined by target-decoy search using the in-house–developed Proline software (http://proline.profiproteomics.fr/). The spectral count metrics was used to rank the proteins and peptides according to their relative abundance in the sample.
Statistical analysis
Differences between groups were assessed using the χ2 or the Fisher’s exact test for qualitative variables and the Mann-Whitney U test for continuous variables. Two-sided P values < .05 were considered statistically significant.
Results and discussion
Patients’ baseline characteristics and IGVL gene usage are shown in supplemental Table 1 (available on the Blood Web site) and Table 1, respectively. Patients with pLCDD were younger (51 years vs 67 years, P = .0001) and more likely to be female (62.5% vs 15.4%, P = .006) compared with patients with pAL amyloidosis. All pLCDD patients had deposits restricted to the lung. Cystic lung disease was the dominant presentation for pLCDD compared with pAL amyloidosis (75.0% vs 15.4%, P = .0005). MS-based proteomic analysis identified immunoglobulin κ peptides as the main protein component of the tissue deposits in each patient. The κ peptides belonging to the constant region were found in 23 (95.8%) and 13 (100%) of patients with pLCDD and pAL amyloidosis, respectively. The combination of APOE, SAP, and APOA4, known as universal amyloid tissue markers, was not found in pLCDD.7,8 We were able to identify peptides from an IGVL gene in 20 (83.3%) patients with pLCDD and in 8 (61.5%) patients with pAL amyloidosis. In pLCDD, IGKV1-8 was the most common IGKVL gene detected (75%), and IGKV1 was the most common κ family detected (86.4%). Peptides from IGKV1-8 were overrepresented in pLCDD compared with pAL amyloidosis (75% vs 11.1%, P = .0033). Furthermore, IGKV1-8 was predominantly associated with a diffuse cystic lung pattern (94%), and it was not found in the 4 pLCDD patients with isolated nodules. In pAL amyloidosis, IGKV1-8 was observed in 1 patient with diffuse cystic lung disease. Imaging, pathological, and proteomic findings of a selected patient with cystic pLCDD are detailed in Figure 1.

Imaging, pathological, and proteomic findings in a 60-year-old man (patient P-08) who underwent bilateral lung transplantation for cysticpLCDD. (A) Computed tomography shows round distinct thin-walled cystic airspaces in both lungs. Light microscopy reveals cysts derived from bronchioles and alveolar spaces (B; original magnification ×0.5, hematoxylin and eosin stain) associated with abundant eosinophilic extracellular amorphous deposits admixed with some plasma cells (C; original magnification ×30, hematoxylin and eosin stain). (D) Electron microscopy shows granular electron-dense deposits in a vessel wall (original magnification ×8000). (E) LC-MS/MS analysis after laser microdissection identifies immunoglobulin κ light chain variable VK1-8 and constant region as the main protein components of these tissue deposits (the proteins are listed according to their relative abundance using spectral count values from Proline software).
Imaging, pathological, and proteomic findings in a 60-year-old man (patient P-08) who underwent bilateral lung transplantation for cysticpLCDD. (A) Computed tomography shows round distinct thin-walled cystic airspaces in both lungs. Light microscopy reveals cysts derived from bronchioles and alveolar spaces (B; original magnification ×0.5, hematoxylin and eosin stain) associated with abundant eosinophilic extracellular amorphous deposits admixed with some plasma cells (C; original magnification ×30, hematoxylin and eosin stain). (D) Electron microscopy shows granular electron-dense deposits in a vessel wall (original magnification ×8000). (E) LC-MS/MS analysis after laser microdissection identifies immunoglobulin κ light chain variable VK1-8 and constant region as the main protein components of these tissue deposits (the proteins are listed according to their relative abundance using spectral count values from Proline software).
The present study confirms that MS-based proteomics is an accurate method for the molecular characterization of light chain deposits in pLCDD. It further highlights that IGKV1-8, a gene rarely used in the normal B-cell repertoire, is significantly associated with pLCDD in its cystic form.
There is little data in the literature about the IGVL gene and light chain family used in LCDD. In the systemic form of LCDD, sequencing of pathogenic light chain in 2 series indicated an overrepresentation of the IGKV4 family and IGKV4-1 subgroup5,12 ; IGVK1-8 was not identified.5 The potential role of the variable region in tissue deposition was documented by showing that amino acid changes in this region are sufficient to promote deposition in the kidney, liver, spleen, and heart of mice expressing a human LCDD IGKV4 chain.13 All of these findings, added to our results, strongly suggest the contribution of the light chain variable domain in organ tropism in LCDD. In the same way, several studies based on polymerase chain reaction sequencing of bone marrow plasma cells support that organ involvement in AL amyloidosis may be partially related to the immunoglobulin light chain repertoire of the clone.14-17 Recently, these results were strengthened with the investigation of IGVL gene usage by LC-MS/MS among 821 cases of AL amyloidosis.11 Interestingly, in this large cohort, IGVL was reported in 14 cases of localized pAL κ amyloidosis.11 IGKV3-20 was the most common IGVL identified (35%), and no IGVL matched with IGKV1-8.11 Beyond the role of immunoglobulin gene usage in organ tropism, the preferential usage of IGKV1-8 in cystic pLCDD strongly upholds the antigen-driven process suggested by the identification of a stereotyped IGHV4-34/IGKV1 receptor expressed by the pulmonary B-cell clone.6 Finally, our study sheds light on the link between IGKV1-8 and diffuse pulmonary cystic disorder, especially LCDD. This raises the question of the IGVL gene implication in the development of this particular pattern.
Limitations of our study include the sensitivity of MS to detect IGVL gene products. There is still room to improve sequence template libraries and peptide-identification algorithms.
In conclusion, we provide evidence that IGKV1-8 is highly associated with cystic pLCDD. This represents an additional feature arguing for a specific entity distinct from the usual systemic form.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Karima Chaoui for technical assistance with MS analyses at Institut de Pharmacologie et de Biologie Structurale.
The work was supported in part by grants from the Région Occitanie, Fonds Européen de Développement Économique et Régional, and the French Ministry of Research (Investissement d’Avenir Program, Proteomics French Infrastructure, ANR-10-INBS-08).
Authorship
Contribution: M. Colombat initiated and supervised the study, performed histological analyses and clinical data collection proteomic analyses, interpreted the data, and wrote the manuscript; M. Camus prepared histologic samples, performed laser microdissection and proteomic analyses, interpreted the data, and wrote the manuscript; S.H., M.-P.C., G.P., H.M., M.S., and M.R.-G. provided tissue samples from patients and carefully read the manuscript; J.G. performed statistical analyses; P.B. carefully read the manuscript and discussed the results; and O.B.-S. provided access to the proteomics infrastructure (especially mass spectrometer instruments) of Institut de Pharmacologie et de Biologie Structurale of Toulouse and carefully read the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Magali Colombat, Département d’Anatomie Pathologique, Institut Universitaire du Cancer, 1 avenue Irène Joliot-Curie, 31059 Toulouse Cedex 09, France; e-mail: colombat.m@chu-toulouse.