Key Points
AAV- and CRISPR/Cas9-mediated gene targeting led to sustained clinically valuable expression of human FIX in a mouse model of hemophilia B.
Because this genome-editing strategy is not mutation position specific, it could be applied to the majority of patients with hemophilia B.

Abstract
Many genetic diseases, including hemophilia, require long-term therapeutic effects. Despite the initial success of liver-directed adeno-associated virus (AAV) gene therapy for hemophilia in clinical trials, long-term sustained therapeutic effects have yet to be seen. One explanation for the gradual decline of efficacy over time is that the nonintegrating AAV vector genome could be lost during cell division during hepatocyte turnover, albeit at a slow pace in adults. Readministering the same vector is challenging as a result of the AAV-neutralizing antibodies elicited by the initial treatment. Here, we investigated the use of clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-mediated homology-directed gene targeting for sustained treatment of hemophilia B. We developed a donor vector containing a promoterless partial human factor IX (FIX) complementary DNA carrying the hyperactive FIX Padua mutation. A single injection of dual AAV vectors in newborn and adult FIX-knockout (FIX-KO) mice led to stable expression of FIX at or above the normal levels for 8 months. Eight weeks after the vector treatment, we subjected a subgroup of newborn and adult treated FIX-KO mice to a two-thirds partial hepatectomy; all of these animals survived the procedure without any complications or interventions. FIX levels persisted at similar levels for 24 weeks after partial hepatectomy, indicating stable genomic targeting. Our results lend support for the use of a CRISPR/Cas9 approach to achieve lifelong expression of therapeutic proteins.
Introduction
Hemophilia B is an X-linked recessive bleeding disorder caused by a defect in the gene encoding coagulation factor IX (FIX). The current treatment for hemophilia B, which entails lifelong IV injections of FIX, is expensive, difficult to adhere to, and not curative. Alternatively, adeno-associated virus (AAV) vector–based gene therapy has the potential to cure the disease through continuous production of FIX. Indeed, AAV gene therapy has shown promise in clinical trials for several diseases,1-8 including hemophilia. However, in most cases, the expression of FIX levels in humans decreases over time.1,8,9 Moreover, long-term therapeutic effects in patients in these trials have yet to be seen. For liver-directed gene therapy, this could be due to the nonintegrating nature of the AAV vector. During hepatocyte turnover, the vector genome could be lost over time, leading to a gradual decline in therapeutic effects. Unfortunately, readministering the vector may not work as a result of the neutralizing antibodies to the capsids elicited from the initial vector administration. One potential solution to this problem is to use genome editing for targeted integration of the therapeutic gene into the host genome.10 This approach was first demonstrated in preclinical models with zinc finger nucleases (ZFNs)11-13 and was subsequently tested ex vivo in the clinic14 and in vivo in patients with hemophilia B or mucopolysaccharidosis.15-17 ZFN-mediated in vivo gene targeting would require cotransduction of 3 vectors in the same cell, which is not very efficient.
We previously developed a dual AAV vector system leveraging the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system. Using homology-directed repair (HDR), we successfully corrected a G-to-A point mutation in 10% of ornithine transcarbamylase (OTC) alleles in the liver of neonatal spfash mice (an OTC deficiency model).18 However, this approach failed in adult mice for 2 reasons: low HDR efficiency in adult mouse liver and large size deletions that extended to the exon, further reducing the residual OTC expression. Another limitation of this approach is that a vector aimed at correcting a specific mutation cannot be applied to other mutations.
Here, we developed a CRISPR/Cas9-mediated gene-targeting approach that could be broadly applied for sustained treatment of patients with hemophilia B. We found that a single injection of a dual AAV8 vector system in neonatal and adult hemophilia B mice achieved stable expression of human FIX (hFIX) with above-normal levels of hFIX activity. Moreover, the treated mice survived a two-thirds partial hepatectomy.
Methods
Plasmid construction
Three 20-nt target sequences preceding a 5′NNGRRT protospacer-adjacent motif sequence located in the 5′ end of exon 2 of murine FIX (mFIX) were selected. These sequences were cloned into pX330.hSaCas9.Puro plasmid.18 The AAV FIX gene-targeting vector contains (1) the U6-FIX single-guide RNA3 (sgRNA3) that specifically targets a region in the 5′ end of exon 2 of mFIX and (2) a codon-optimized partial hFIX complementary DNA (cDNA) sequence spanning the remaining exon 2-8 that carries the hyperactive Padua mutation and the bovine growth hormone polyA flanked by 0.9-kb homology arms on each side (Figure 1). The “untargeted” AAV.control.donor lacks the protospacer sequence from the U6-FIX sgRNA3 cassette. All plasmid constructs were verified by sequencing. The Cas9 from Staphylococcus aureus (SaCas9) expression vector AAV.TBG.SaCas9 has been described previously.18
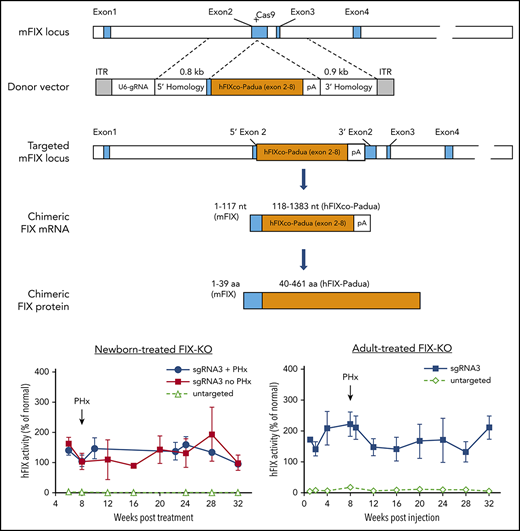
![Figure 1. In vivo gene targeting of the FIX locus in FIX-KO mouse liver by AAV.SaCas9. Schematic diagrams of the mouse FIX locus show the SaCas9.sgRNA3 target site located in the 5′ end of exon 2; the AAV donor vector that contains U6-sgRNA3 and partial cDNA of codon-optimized, Padua-containing hFIX cDNA (hFIXco-Padua [exon 2-8]) followed by a polyA and flanked by homology arms; the modified mFIX locus after homologous recombination; the chimeric FIX mRNA transcribed from the gene-targeted FIX locus; and the translated chimeric FIX protein. Blue boxes indicate mFIX exons or exon-derived mRNA or protein sequences, and orange boxes indicate codon-optimized hFIX cDNA (exon 2-8), mRNA, or protein.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/26/10.1182_blood.2019000790/3/m_bloodbld2019000790f1.png?Expires=1764378344&Signature=LgRp3ZAJVoC48vlwmdDLdmbSfilSjOyZIn02~kn2sxz3D2otZLEdb79SzHKE-R3sn1Av~PC3neisSV9IC9WzNUvPeyfNpD8RgluoxALDETQPukQ4gHjfUm2xMIQEobOxI0sv625c8sBPwN-fbkNFdGTyHWvs8~F0sIhOfwsoS00-A0vpwl7kUVFqr4LNHmiwFjIilCVXgNH4lC5OgKUFmtSCeERKvfUPDFZSO7v0ewm73X3ItP7hb1k9kWRgM4t1SPY3qZTR-jPVwm02b2klFB2tlL5-pb-TogmJl6YCaIV6iIqKYu4W1ZylDkceRK3CiTRpJ3e7QUBL56gfL2dxRA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
In vivo gene targeting of the FIX locus in FIX-KO mouse liver by AAV.SaCas9. Schematic diagrams of the mouse FIX locus show the SaCas9.sgRNA3 target site located in the 5′ end of exon 2; the AAV donor vector that contains U6-sgRNA3 and partial cDNA of codon-optimized, Padua-containing hFIX cDNA (hFIXco-Padua [exon 2-8]) followed by a polyA and flanked by homology arms; the modified mFIX locus after homologous recombination; the chimeric FIX mRNA transcribed from the gene-targeted FIX locus; and the translated chimeric FIX protein. Blue boxes indicate mFIX exons or exon-derived mRNA or protein sequences, and orange boxes indicate codon-optimized hFIX cDNA (exon 2-8), mRNA, or protein.
In vivo gene targeting of the FIX locus in FIX-KO mouse liver by AAV.SaCas9. Schematic diagrams of the mouse FIX locus show the SaCas9.sgRNA3 target site located in the 5′ end of exon 2; the AAV donor vector that contains U6-sgRNA3 and partial cDNA of codon-optimized, Padua-containing hFIX cDNA (hFIXco-Padua [exon 2-8]) followed by a polyA and flanked by homology arms; the modified mFIX locus after homologous recombination; the chimeric FIX mRNA transcribed from the gene-targeted FIX locus; and the translated chimeric FIX protein. Blue boxes indicate mFIX exons or exon-derived mRNA or protein sequences, and orange boxes indicate codon-optimized hFIX cDNA (exon 2-8), mRNA, or protein.
AAV vector production
All AAV8 vectors were produced by the Penn Vector Core at the University of Pennsylvania (UPenn), as previously described.19 The genome titer (genome copies [GCs] per milliliter) of AAV vectors was determined by digital droplet polymerase chain reaction (PCR), which is typically twofold to threefold higher than the titer determined by quantitative PCR (qPCR).20 All vectors used in this study passed an endotoxin assay using the QCL-1000 Chromogenic LAL kit (Cambrex Bio Science, Walkersville, MD).
Cell culture, transfection, and GUIDE-seq
H2.35 (ATCC CRL-1995) Mus musculus liver cells were maintained in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum and cultured at 37°C with 5% CO2. For target validation, plasmids expressing SaCas9 and sgRNA were transfected into H2.35 cells using Lipofectamine LTX with Plus reagent (Thermo Fisher Scientific, Waltham, MA), per the manufacturer’s recommendations. A cell-based genome-wide unbiased identification of double-strand breaks enabled by sequencing (GUIDE-seq) experiment was performed by cotransfecting the plasmid expressing SaCas9 and sgRNA3 and double-stranded oligodeoxynucleotides into H2.35 cells, as described.21
Animal studies
FIX-knockout (FIX-KO) mice,22 which lack the last C-terminal 164 amino acids of the FIX protein and the 3′ untranslated region, were maintained in an Association for Assessment and Accreditation of Laboratory Animal Care–accredited and Public Health Service–assured facility at UPenn. All animal procedures were performed in accordance with the Institutional Animal Care and Use Committee of UPenn. Mating cages were monitored daily for births. Newborn postnatal day 2 male pups received a temporal vein injection of a mixture of 2 vectors at a volume of 50 µL, as described.23 Plasma samples for hFIX assays were obtained by retro-orbital bleeding 6 weeks postvector treatment and every 2 to 4 weeks thereafter. In the adult mouse study, 8- to 10-week-old FIX-KO male mice received a mixture of 2 vectors via the tail vein. Mice were bled at various time points starting 1 week postinjection. Mice were euthanized 32 weeks postinjection. A two-thirds partial hepatectomy was performed on a subset of targeting vector–treated FIX-KO mice 8 weeks postvector injection, as described.24 A subset of mice treated with the control vector was euthanized at 8 weeks postvector injection, and liver tissues were harvested as controls.
FIX assays
hFIX protein levels in the plasma were measured by enzyme-linked immunosorbent assay (ELISA)25 and are shown as a percentage of Pooled Normal Human Plasma (George King Bio-Medical, Overland Park, KS), which we included in each assay as a reference. FIX activity was measured by 1-step activated partial thromboplastin time (aPTT) using a Stago STart Hemostasis Analyzer (Diagnostica Stago, Parsippany, NJ), as previously described with modifications.22 A standard curve was generated using Factor Assay Control Plasma in FIX-deficient plasma (both from George King Bio-Medical). Factor Assay Control Plasma was calibrated using a reference standard (the Fourth World Health Organization International Standard NIBSC code 07/182). Samples were compared with the standard curve to obtain the relative activity of hFIX. Samples were diluted twofold to eightfold in FIX-deficient plasma to obtain levels within the interpretable range. Untreated FIX-KO mice typically showed <2% of normal activity levels.
Quantification of chimeric murine-hFIX mRNA
RNA was isolated from liver using TRIzol Reagent and reverse transcribed using a High-Capacity cDNA Reverse Transcription Kit (both from Thermo Fisher Scientific). Real-time PCR was performed to measure hybrid murine-hFIX (m-hFIX) messenger RNA (mRNA) using forward primer CCCGGCTCTCATCACCATCT, reverse primer GTTGCCCTGCACGAACTCTT, and probe FAM-GGCCCAAGCGGTACAACAGCGGC-MGB. Transgene expression levels were normalized to GAPDH levels (Thermo Fisher Scientific).
Indel analysis on PCR amplicons
In vitro validation of each mFIX sgRNA was performed on genomic DNA extracted from transfected H2.35 cells by a SURVEYOR nuclease assay (Integrated DNA Technologies, Coralville, IA), as described,18,26 using PCR primers listed in supplemental Table 1 (available on the Blood Web site). In vivo genome-editing efficiency was measured on genomic DNA extracted from mouse liver tissues 8 and 32 weeks following vector administration. This was used as a template for nested PCR amplifications using Q5 High-Fidelity DNA Polymerase (NEB, Ipswich, MA) with primers flanking the on- and off-target loci (supplemental Table 1). Insertion and deletion (indel) analyses were performed on deep sequencing of the PCR amplicons, as previously described.27
Statistical analyses
Statistical analyses were performed with GraphPad Prism 7 for Windows. One-way analysis of variance and Dunnett’s multiple-comparisons test were used to compare variables with a single control. To compare untargeted and targeted groups, the nonparametric Mann-Whitney U test (2-tailed) was used. Because of the relatively small sample size, normality testing was not feasible. Group averages are presented as mean ± standard deviation (SD).
Results
CRISPR/Cas9-mediated gene-targeting vector for hemophilia B mice
We aimed to develop an HDR-mediated gene-targeting vector for hemophilia B mice. We selected exon 2 of the mFIX gene as the targeting site to avoid promoter activity in the 5′ untranslated region of the 5′ HDR arm. We searched for protospacer-adjacent motif sequences (NNGRRT) in the 5′ end of exon 2 and identified potential 20-nt protospacer sequences for SaCas9. We further evaluated 3 sgRNA sequences, sgRNA1-3 (supplemental Table 1), after transfecting plasmids expressing SaCas9 and individual sgRNAs into a mouse H2.35 cell line. We obtained evidence of double-strand breaks (DSBs) and indels at the desired site for all 3 sgRNAs using SURVEYOR (supplemental Figure 1). sgRNA3 showed slightly higher on-target editing efficiency than the other 2 sgRNAs.
We next developed an AAV8 gene-targeting donor vector that contains (1) sgRNA3 driven by the U6 promoter to target exon2 of mFIX and (2) a codon-optimized partial hFIX cDNA sequence spanning the remaining exon 2 to exon 8 and carrying the hyperactive Padua mutation (hFIXco-Padua), followed by the bovine growth hormone polyA, flanked by 0.8-kb and 0.9-kb homology arms on the 5′ and 3′ sides, respectively (Figure 1). The untargeted control donor vector contains all components, with the exception of the 20-nt target sequence of the sgRNA (referred to as the AAV8.untargeted donor). Following CRISPR/Cas9-mediated HDR, the partial hFIXco cDNA should be fused with the 5′ end of mFIX exon 2. This would lead to the transcription of a chimeric m-hFIX mRNA by the native mFIX promoter, in which the first 117 nt come from mFIX and rest of the nucleotides (118-1383) come from hFIXco-Padua (Figure 1). In the translated chimeric FIX protein, the first 39 amino acids derive from mFIX, and the rest of the protein sequences (40-461) derive from hFIX-Padua (Figure 1).
Efficacy of in vivo gene targeting in neonatal FIX-KO mouse liver by dual AAV.SaCas9 gene-targeting vectors
To determine the efficacy of in vivo gene targeting in neonatal hemophilia B mice, we coinjected AAV8.SaCas9 (5 × 1010 GCs per pup) and AAV8.sgRNA3.hFIXco-Padua donor (2.5 × 1011 GCs per pup) into postnatal day 2 FIX-KO male pups via the temporal vein. At 6 weeks postinjection, we collected plasma to measure hFIX protein and activity levels. All treated mice showed therapeutic expression levels of hFIX protein (10.9% ± 1.6% of normal, n = 6) and above-normal levels of activity (147.3% ± 21.0% of normal, n = 6) owing to the hyperactive FIX-Padua variant incorporated into the donor vector (Figure 2A-B). We performed a two-thirds partial hepatectomy on a subset of the treated mice at 8 weeks postinjection. All 3 mice survived the procedure without any complications or interventions, such as experiencing excessive bleeding and/or requiring infusion with clotting factors. hFIX expression and activity levels remained stable until the end of the study (week 32) and were similar to mice without partial hepatectomy (Figure 2A-B). Mice that received coinjected SaCas9 vector and control donor vector had hFIX protein and activity levels close to baseline (Figure 2A-B). One control vector–treated mouse died <8 weeks postinjection from internal bleeding. We euthanized 3 vector-treated mice at 8 weeks postinjection and harvested liver samples to serve as controls.
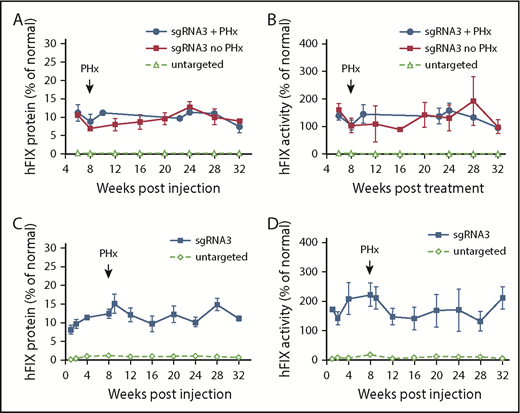
Efficacy of AAV8.SaCas9-mediated hepatic gene targeting in FIX-KO mice treated as newborns and adults. (A) hFIX protein levels in FIX-KO mouse plasma after neonatal temporal vein injection of AAV8.SaCas9 (5 × 1010 GCs per pup) and AAV8.sgRNA3.hFIXco-Padua donor (2.5 × 1011 GCs per pup) (n = 6). A subset of treated mice (n = 3) was subjected to two-thirds partial hepatectomy 8 weeks postvector treatment. All treated mice were euthanized 32 weeks postvector treatment. Untargeted FIX-KO pups (n = 8) received AAV8.SaCas9 (2.5 × 1011 GCs per pup) and AAV8.control.hFIXco-Padua donor (2.5 × 1012 GCs per pup). One untargeted mouse died at week 8 from hemorrhage, 3 were euthanized at week 8 for analyses, and the remaining 4 were euthanized at 32 weeks. (B) hFIX activity in FIX-KO mouse plasma after neonatal vector treatment. (C) hFIX protein levels in adult-treated FIX-KO mouse plasma after tail vein injection of AAV8.SaCas9 (5 × 1011 GCs per mouse) and AAV8.sgRNA3.hFIXco-Padua donor (5 × 1012 GCs per mouse) (n = 5). All treated mice were subjected to two-thirds partial hepatectomy 8 weeks postvector treatment and were euthanized 32 weeks postvector treatment. Untargeted FIX-KO mice (n = 8) received AAV8.SaCas9 (5 × 1011 GCs per mouse) and AAV8.control.hFIXco-Padua donor (5 × 1012 GCs per mouse). Three of the untargeted mice were euthanized at week 8, and liver samples were harvested for analyses; the remaining 5 mice were euthanized at 32 weeks. (D) hFIX activity in adult-treated FIX-KO mouse plasma. Data are mean ± SD.
Efficacy of AAV8.SaCas9-mediated hepatic gene targeting in FIX-KO mice treated as newborns and adults. (A) hFIX protein levels in FIX-KO mouse plasma after neonatal temporal vein injection of AAV8.SaCas9 (5 × 1010 GCs per pup) and AAV8.sgRNA3.hFIXco-Padua donor (2.5 × 1011 GCs per pup) (n = 6). A subset of treated mice (n = 3) was subjected to two-thirds partial hepatectomy 8 weeks postvector treatment. All treated mice were euthanized 32 weeks postvector treatment. Untargeted FIX-KO pups (n = 8) received AAV8.SaCas9 (2.5 × 1011 GCs per pup) and AAV8.control.hFIXco-Padua donor (2.5 × 1012 GCs per pup). One untargeted mouse died at week 8 from hemorrhage, 3 were euthanized at week 8 for analyses, and the remaining 4 were euthanized at 32 weeks. (B) hFIX activity in FIX-KO mouse plasma after neonatal vector treatment. (C) hFIX protein levels in adult-treated FIX-KO mouse plasma after tail vein injection of AAV8.SaCas9 (5 × 1011 GCs per mouse) and AAV8.sgRNA3.hFIXco-Padua donor (5 × 1012 GCs per mouse) (n = 5). All treated mice were subjected to two-thirds partial hepatectomy 8 weeks postvector treatment and were euthanized 32 weeks postvector treatment. Untargeted FIX-KO mice (n = 8) received AAV8.SaCas9 (5 × 1011 GCs per mouse) and AAV8.control.hFIXco-Padua donor (5 × 1012 GCs per mouse). Three of the untargeted mice were euthanized at week 8, and liver samples were harvested for analyses; the remaining 5 mice were euthanized at 32 weeks. (D) hFIX activity in adult-treated FIX-KO mouse plasma. Data are mean ± SD.
To evaluate the vector dose effects on in vivo gene targeting in neonatal mice, we increased the vector dose for AAV8.SaCas9 by fivefold, in combination with increased donor vector doses (supplemental Figure 3). The increments of hFIX protein expression levels (supplemental Figure 3A) and activity levels (supplemental Figure 3B) were insignificant, with the exception of mice treated with a 10-fold higher donor vector dose (2.5 × 1012 GCs per pup).
Efficacy of in vivo gene targeting in adult FIX-KO mouse liver by dual AAV.SaCas9 gene-targeting vectors
To evaluate the efficacy of in vivo gene targeting in adult hemophilia B mice, we coinjected AAV8.SaCas9 (5 × 1011 GCs per mouse) and AAV8.sgRNA3.hFIXco-Padua donor (5 × 1012 GCs per mouse) into 8- to 10-week-old male FIX-KO mice via the tail vein. Eight weeks later, all animals were subjected to a two-thirds partial hepatectomy with no adverse consequences. At 1 week postinjection, we measured hFIX protein and activity levels in the plasma. All treated mice expressed therapeutic levels of hFIX protein (8.2% ± 1.2% of normal, n = 5) and above-normal levels of activity (171.6% ± 5.2% of normal, n = 5) (Figure 2C-D). Levels increased slightly by week 4 postinjection and stabilized until the end of the study (week 32). Partial hepatectomy had no impact on expression. Mice that received coinjected SaCas9 vector and control donor vector showed very low levels of hFIX protein and activity during the first 2 weeks, which then increased to 1.1% ± 0.2% of normal and 5.9% ± 3.0% of normal, respectively (week 4, n = 8) (Figure 2C-D). We euthanized 3 vector-treated mice at 8 weeks postinjection and the remaining 5 mice at 32 weeks postinjection and harvested liver samples as controls.
We also performed a dose-response study in adult mice to evaluate the effects of vector dose on in vivo gene targeting. As shown in supplemental Figure 4, increasing the donor vector dose by threefold, from 5 × 1012 GCs per mouse to 1.5 × 1013 GCs per mouse, improved hFIX protein levels by 1.8-fold and hFIX activity by 1.7-fold; conversely, decreasing the donor vector dose by 10-fold, to 5 × 1011 GCs per mouse, reduced hFIX protein and activity levels by 1.7- and 1.6-fold (95% of normal activity), respectively. Reducing the donor vector dose by threefold (1.5 × 1012 GCs per mouse) resulted in a minor reduction in hFIX expression and activity. A further reduction in the AAV8.SaCas9 vector by fivefold, to 1 × 1011 GCs per mouse, and keeping the donor vector fixed at 1.5 × 1012 GCs per mouse resulted in a 1.5-fold reduction in FIX expression.
Expression of hFIX in live mice following treatment with gene-targeting vector
We measured chimeric m-hFIX mRNA copies in liver harvested at 8 and 32 weeks postinjection by reverse transcription, followed by qPCR using primers spanning the junction of mFIX and hFIX. We detected high levels of the chimeric mRNA but did not observe a difference in these levels between weeks 8 and 32 in any of the groups (Figure 3). Untargeted vector-treated mice showed baseline levels.
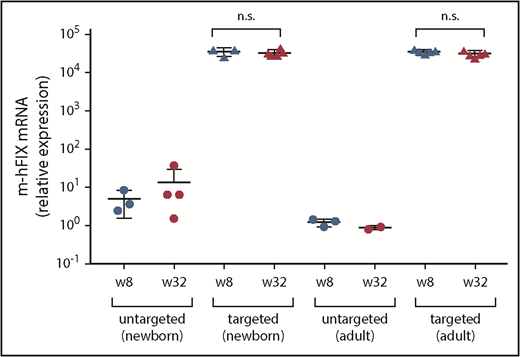
Transgene expression in liver samples collected at 8 and 32 weeks after treatment with dual vectors for gene targeting as neonates or adults. Chimeric m-hFIX mRNA levels in liver measured by reverse-transcription qPCR using primers/probe spanning the junction of murine and hFIX cDNA. Liver samples were collected from targeting vector–treated FIX-KO mice at 8 weeks after vector treatment via two-thirds partial hepatectomy and at 32 weeks during necropsy. Untargeted control mice were euthanized at 8 and 32 weeks after vector treatment for liver collection. Each circle represents an individual mouse. Data are mean ± SD. A 2-tailed Mann-Whitney U test was used to compare m-hFIX mRNA levels at weeks 8 and 32. n.s., not statistically significant.
Transgene expression in liver samples collected at 8 and 32 weeks after treatment with dual vectors for gene targeting as neonates or adults. Chimeric m-hFIX mRNA levels in liver measured by reverse-transcription qPCR using primers/probe spanning the junction of murine and hFIX cDNA. Liver samples were collected from targeting vector–treated FIX-KO mice at 8 weeks after vector treatment via two-thirds partial hepatectomy and at 32 weeks during necropsy. Untargeted control mice were euthanized at 8 and 32 weeks after vector treatment for liver collection. Each circle represents an individual mouse. Data are mean ± SD. A 2-tailed Mann-Whitney U test was used to compare m-hFIX mRNA levels at weeks 8 and 32. n.s., not statistically significant.
On-target indel frequency and HDR-mediated gene-targeting efficiency
We analyzed on-target indel frequency in DNA isolated from FIX-KO mice at 8 and 32 weeks postvector treatment by deep sequencing of PCR amplicons of the targeted mFIX locus. Mice treated with targeting vector as neonates (AAV8.SaCas9 [5 × 1010 GCs per pup] and AAV8.sgRNA3.hFIXco-Padua donor [2.5 × 1011 GCs per pup]) had a mean indel frequency of 42% (range, 38-47%, n = 3) at week 8 and 36% (range, 30-42%, n = 6) at week 32 (Figure 4A). Untargeted mice had background levels of 0.2% and 0.3% at 8 and 32 weeks, respectively, similar to untreated mice (Figure 4A). Mice treated with targeting vector as adults (AAV8.SaCas9 [5 × 1011 GCs per mouse] and AAV8.sgRNA3.hFIXco-Padua donor [5 × 1012 GCs per mouse]) had a mean indel frequency of 37% (range, 30-45%, n = 5) at week 8 and 36% (range, 34-39%, n = 5) at week 32 (Figure 4B). The untargeted mice had background levels of 0.3% (n = 3) at 8 weeks and a slightly higher indel frequency of 1.4% (n = 5) at 32 weeks (Figure 4B).
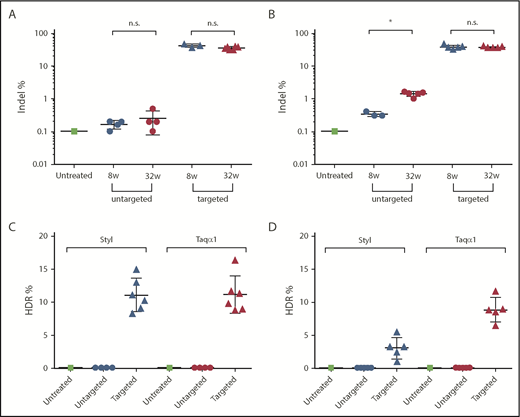
Indel and HDR-mediated gene-targeting efficiency analyses. Liver DNA was isolated from FIX-KO mice at 8 and 32 weeks posttreatment with dual gene–targeting vectors or untargeted vectors. DNA from an untreated FIX-KO mouse served as control. Indel analysis on the targeted mFIX locus was performed by deep sequencing on liver DNA isolated at 8 and 32 weeks posttreatment. HDR-mediated gene-targeting efficiency was analyzed by LMU-PCR, following digestion with StyI or TaqαI (see supplemental Figure 5), on liver DNA isolated at 32 weeks posttreatment. Indel frequency in mice treated as neonates (A) or as adults (B). HDR frequency in mice treated as neonates (C) or as adults (D). Each data point represents an individual mouse. Data are mean ± SD. *P < .05, 2-tailed Mann-Whitney U test. n.s., not statistically significant.
Indel and HDR-mediated gene-targeting efficiency analyses. Liver DNA was isolated from FIX-KO mice at 8 and 32 weeks posttreatment with dual gene–targeting vectors or untargeted vectors. DNA from an untreated FIX-KO mouse served as control. Indel analysis on the targeted mFIX locus was performed by deep sequencing on liver DNA isolated at 8 and 32 weeks posttreatment. HDR-mediated gene-targeting efficiency was analyzed by LMU-PCR, following digestion with StyI or TaqαI (see supplemental Figure 5), on liver DNA isolated at 32 weeks posttreatment. Indel frequency in mice treated as neonates (A) or as adults (B). HDR frequency in mice treated as neonates (C) or as adults (D). Each data point represents an individual mouse. Data are mean ± SD. *P < .05, 2-tailed Mann-Whitney U test. n.s., not statistically significant.
To characterize the editing events in the targeted locus and to estimate HDR-mediated gene-targeting efficiency, we developed a method called ligation-mediated PCR (LMU-PCR) and coupled it with unique molecular indices, followed by deep sequencing (supplemental Figure 5). We first digested the genomic DNA isolated at week 32 postvector injection with Taqα1 or StyI, which have recognition sites in the homology arms and in the donor vector (supplemental Figure 5A-B). Subsequently, we performed LMU-PCR, which generated relatively similar sizes of PCR amplicons from untargeted and targeted FIX loci (supplemental Figure 5C-D). Following deep sequencing of the nested PCR amplicons, we analyzed the mapped reads to calculate the ratio of reads containing the expected hFIXco sequence/total mapped reads, and defined it as HDR-mediated gene-targeting efficiency. The targeted mice treated as neonates showed a mean targeting efficiency of 11% by StyI and Taqα1 assays (range, 8-15% for StyI; range, 9-16% for Taqα1; n = 6; Figure 4C). The untargeted mice (n = 4) showed background levels (0.02%) by both assays (Figure 4C). The targeted mice treated as adults showed a mean targeting efficiency of 3% by the StyI assay (range, 1-6%; n = 6) and 9% by the Taqα1 assay (range, 7-12%; n = 5) (Figure 4D). The untargeted mice (n = 5) showed background levels by both assays (0.07% by StyI and 0.05% by Taqα1) (Figure 4D). The reasons for the discrepancies in the HDR efficiency in adult-treated mice by the StyI and Taqα1 assays are unclear.
Detailed analysis of the sequences obtained by LMU-PCR revealed a more complex pattern in the nature of the target region after genome editing. In addition to the sequences corresponding to parental genomic DNA and HDR-mediated insertion of the transgene, we found sequences mapping to different elements of the AAV.SaCas9 and donor vectors, such as the promoter, polyA, transgene, and inverted terminal repeats (ITRs), indicative of nonhomologous end joining (NHEJ)-mediated insertions (supplemental Figure 6).
Off-target activity in vitro and in vivo
We first performed GUIDE-seq21 in the H2.35 mouse cell line to evaluate the off-target activity of sgRNA3 in the mouse genome. We did not detect any off-target sites, despite efficient on-target editing (supplemental Figure 2). However, off-target activity can still occur in vivo after administering the gene-targeting vectors, because the doses of the nuclease and sgRNA, as well as the time of exposure, are different from those used in vitro. Therefore, we designed primers encompassing 10 of the most probable off-target loci for sgRNA3, as predicted by the online tool Benchling (http://www.benchling.com; supplemental Tables 1 and 2). We generated amplicons from liver DNA isolated at 32 weeks postvector treatment and then quantified the indels by next-generation sequencing (NGS) in the predicted off-target regions. We observed similar low indel percentages in these loci from mice treated with targeted or untargeted vectors compared with the on-target indel percentage in livers administered with targeted, but not untargeted, vectors (supplemental Table 3), suggesting that sgRNA3 is highly specific for its intended target sequence, similar to what we observed in vitro.
Discussion
In this study, we used an AAV-delivered CRISPR/Cas9 approach to stably express hFIX in neonatal and adult hemophilia B mice. In treated animals, circulating FIX protein levels ranged from 10% to 15% of normal, and FIX activities were above the normal levels (120-160%). Moreover, we demonstrated clinical efficacy in treated mice that were subjected to a two-thirds partial hepatectomy, a major invasive procedure for a normal mouse.
Several research groups have used AAV-mediated gene targeting to achieve therapeutic levels of gene expression in mouse models, leveraging different targeting loci.11-13,28,29 Studies using ZFN targeted intron 1 of the albumin gene to take advantage of the strong albumin promoter.11-13 The CRISPR FIX study targeted intron 1 of the mFIX gene to use the endogenous mFIX promoter.29 Finally, an approach without nuclease targeted the last exon of albumin right before the stop codon, enabling a preceding 2A-peptide coding sequence to be integrated just upstream from the albumin stop codon.28 In the present study, we targeted the 5′ end of exon 2 of mFIX to use the endogenous mFIX promoter. This is similar to Ohmori et al’s CRISPR FIX study, although they targeted the intron 1 region of the mFIX gene.29 In addition, we incorporated the FIX hyperactive Padua mutation into the codon-optimized hFIX partial cDNA (3′ exon 2–exon 8) in the donor vector.30 Thus, this strategy could be applied to the majority of hemophilia B patients, with the exception of those with mutations in the FIX promoter region or in the 5′ end of the FIX coding sequence (before the sgRNA cleavage site).31 The Padua mutation significantly improved the efficacy of gene targeting in hemophilia B mice; however, hyperactive mutations have rarely been identified for other disease genes. Therefore, this strategy cannot be widely applied to other diseases. The Padua effect that we reported here is ∼15-fold, and it is higher than other investigators have reported (approximately eightfold).30 One possible reason is the different reference standards used for the hFIX ELISA and aPTT assays. Although the reference standard we used in the aPTT assay was calibrated with an international reference standard, the reference for hFIX antigen levels was just pooled normal plasma, because there is no established international reference standard for hFIX antigen.
In addition to AAV-mediated gene targeting, an all-in-1 single adenoviral vector containing the Tet-on-inducible CRISPR/Cas9, guide RNA, and donor DNA reportedly achieved higher HDR efficiency than a 2-vector system in an in vitro model for canine hemophilia B.32 However, the strong immunity associated with adenoviral vectors may limit its in vivo application.
The targeting efficiencies in adult-treated mice were much higher than expected based on our previous in vivo genome-editing studies in OTC spfash mice.18 HDR-based mutation correction via CRISPR/Cas9 was efficient in the liver of OTC spfash mice treated as newborns, but not adults, likely because HDR primarily occurs during cell proliferation (S/G2 phase of the cell cycle).33
Because of the large homology arms (800-900 bp at each end) of the donor vector, there is no straightforward way to measure HDR-mediated gene-targeting efficiency. Therefore, we developed a method, LMU-PCR, to quantify the levels of insertion of the hFIXco sequences. Assays with 2 restriction enzymes showed a similar targeting efficiency of 11% to 13% in mice treated as neonates (Figure 4C). In adult-treated mouse liver, the Taqα1 assay showed threefold higher HDR efficiency than the StyI assay (3%) (Figure 4). Because we did not observe this in neonatal-treated mice, it is possible that the high amount of donor vector genomes that persisted in the adult-treated liver interfered with LMU-PCR. Detailed analysis of the sequences obtained by LMU-PCR revealed a complex pattern of genome structure around the target region (supplemental Figure 6). In addition to the sequences corresponding to parental genomic DNA and HDR-mediated insertion of the hFIXco, we found different parts of the AAV vector sequences inserted into DSBs, including partial portions of the promoter, polyA, transgene, and AAV ITRs. Some of these insertions may be too large to be captured by short PCR amplicon–NGS; therefore, short PCR amplicon–NGS likely underestimates the true indel frequencies. When delivered by AAV vectors, insertion of AAV sequences into the Cas9 cleavage site is expected, because AAV vector sequences can be integrated into the genome after induction of DSBs in the cell genome.34 Furthermore, AAV ITR sequences were detected after treatment of cells with a ZFN targeting to the AAVS1 locus.35 These studies indicate that insertion of AAV sequences by NHEJ is an expected secondary effect of any AAV-delivered genome-editing approach.
We cannot determine the extent of the AAV sequences integrated into the target region as a result of limitations of the NGS amplicons; however, some hFIX expression could be derived from the donor vector inserted by NHEJ rather than HDR. Studies using ZFNs have reported NHEJ-mediated integration of the gene-targeting vector.12,13 The duplication of the homology arms following NHEJ-mediated insertion of donor vector DNA may cause recombination between the duplicate HDR arms, eventually resulting in a sequence similar to HDR-mediated gene targeting (Figure 4A).
Off-target activity is a major safety concern for genome editing. A recent study showed that appropriately designed guide RNAs can direct efficient in vivo editing in mouse livers with no detectable off-target mutations.36 We evaluated the specificity of sgRNA3 in 2 ways: using GUIDE-seq in a mouse cell line and using NGS validation on the top 10 potential off-target sites identified by a computer algorithm in liver DNA from treated animals. Despite efficient on-target editing, we did not detect any off-target activity, indicating high specificity of sgRNA3 (supplemental Figure 2; supplemental Table 3).
Although our gene-targeting strategy achieved stable and complete normalization of hemostasis in the hemophilia B mice, it should be noted that the vector dose used in the gene-target study is significantly higher than those used in AAV gene-therapy studies. In adult hemophilia B mice, a single injection of AAV8.TBG.hFIXco-Padua at a 3-log lower dose (1 × 109 GCs per mouse) could achieve similar FIX activity as in this study (L.W., unpublished data).
In conclusion, we have demonstrated a therapeutic effect of AAV-delivered CRISPR/Cas9-mediated gene targeting in a mouse model of hemophilia B. A single injection of dual AAV gene-targeting vectors in neonatal and adult mice achieved robust and sustained expression of hFIX that was clinically beneficial. This genome-editing strategy is not mutation position specific and could be broadly applied to majority of patients with the same disease.
For original data, please contact liliwang@upenn.edu. Data supporting the findings of this study are available within the paper and its supplementary data. The original deep-sequencing data are available at NCBI BioProject under accession code PRJNA526239.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the following UPenn services: the Penn Vector Core at UPenn for supplying AAV vectors, the Nucleic Acid Technologies Core of the Gene Therapy Program for assistance with deep sequencing, and the Program for Comparative Medicine for mouse procedures.
This work was supported by Penn Medicine.
Authorship
Contribution: L.W., Y.Y., and J.M.W. conceived this study and designed the experiments; Y.Y. constructed the donor vectors; D.M., J.W., Y.Y., and L.W. performed mouse studies; Y.Y., C.A.B., J.Z., A.S., C.L., and Z.H. performed indel and targeting efficiency analyses; A.S. and C.L. performed GUIDE-seq analysis; Y.C. and M.L. conducted bioinformatics analyses of the deep-sequencing data and statistical analyses; J.W. performed the hFIX ELISA and aPTT assays; Z.H. performed reverse-transcription qPCR analysis; L.W. wrote the manuscript; J.M.W., C.A.B., and J.Z. edited the manuscript; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: J.M.W. is an advisor to, holds equity in, and has a sponsored research agreement with Scout Bio and Passage Bio; and he has sponsored research agreements with Ultragenyx, Biogen, Janssen Pharmaceuticals, Precision BioSciences, Moderna Therapeutics, and Amicus Therapeutics, who are licensees of Penn technology. J.M.W., L.W., and Y.Y. are inventors on patents that have been licensed to various biopharmaceutical companies. The remaining authors declare no competing financial interests.
The current affiliation for J.Z. is Sarepta Therapeutics, Burlington, MA.
Correspondence: James M. Wilson, Gene Therapy Program, Department of Medicine, University of Pennsylvania, 125 South 31st St, Suite 1200, Philadelphia, PA 19104; e-mail: wilsonjm@upenn.edu.